When use epw, I find it's difficult to set the the 'projections' in inputs. Where can I get the projections such as 'f=-0.125,-0.125, 0.375:s' ?
And can I get the right results using the proj(1)= 'random' in the calculation? Thanks!
About the projections
Moderator: stiwari
-
fenglei2000
- Posts: 1
- Joined: Tue Mar 23, 2021 9:10 am
- Affiliation: North China University of Science
Re: About the projections
Dear fenglei2000:
Your question is about the Wannierization and regarding the projections, you need to read the section of II. 3 "Projections" of the user guide at http://www.wannier.org/support/ .
Regarding your second question:
Generally speaking, instead of choosing the random projections, it is better to choose the initial projections which are suitable to your material.
Sincerely,
H. Lee
Your question is about the Wannierization and regarding the projections, you need to read the section of II. 3 "Projections" of the user guide at http://www.wannier.org/support/ .
Regarding your second question:
Generally speaking, instead of choosing the random projections, it is better to choose the initial projections which are suitable to your material.
Sincerely,
H. Lee
Re: About the projections
Dear H. Lee
I don't really know what is initial projections, i have also met some problems.when i plot pentacene's MLWFs using Gaussian cube format, it should be localized on the melecular stucture cause i choose VB and VB-1 bands to construct MLWFs, so it is logically should be like homo and homo-1 in
Gaussian's result. However, my result is not like that. Here is an image of MLWFs: [img][https://i.loli.net/2021/05/18/ItgQnyX4zaVqukC.png]
Here is my band structure:[img][https://i.loli.net/2021/05/18/eJUycStPOqZo8m1.png], i choose two bands just marked in image. And i've tried wannier_plot_mode = molecule.But it is not good.
Here is the pdos: [img][https://i.loli.net/2021/05/18/i4YQ8jdqgRDoNXs.png], it is known that C's py orbital is the main contribution to VB and VB-1.
And here are my .win file:
---------------------------------------
exclude_bands = 1:82,85:150
num_wann = 2
num_bands = 2
num_iter = 500
!dis_win_max = 2
!dis_win_min = 2
!dis_froz_max = 1
!dis_froz_min = 1
!dis_num_iter = 50
!dis_mix_ratio = 1.0
kmesh_tol = 0.00001
write_hr= .true.
write_hr_diag=.true.
wannier_plot= .true.
wannier_plot_format = cube
wannier_plot_mode = crystal
wannier_plot_radius = 5
wannier_plot_scale = 1.0
wannier_plot_supercell = 3
wannier_plot_list = 1-2
!bands_plot =.true.
restart = plot
write_xyz=.TRUE.
!gamma_only=.TRUE.
begin atoms_frac
C 0.12959 0.03292 0.39138
C 0.88207 0.98320 0.61377
C 0.15861 0.15508 0.32454
C 0.85321 0.86107 0.68068
C 0.09267 0.07521 0.20891
C 1.91916 0.94108 0.79630
C 0.12093 0.19586 0.13924
C 0.89109 0.82037 0.86600
C 0.05548 0.11521 0.02537
C 0.95656 0.90105 0.97985
C 0.92886 0.78029 0.05088
C 0.08311 0.23596 0.95430
C 0.99391 0.86145 0.16347
C 0.01777 0.15492 0.84169
C 0.96630 0.74018 0.23612
C 0.04512 0.27619 0.76894
C 0.03194 0.82313 0.34656
C 0.97947 0.19315 0.65851
C 0.54261 0.56482 0.37485
C 0.46850 0.45148 0.62262
C 0.48052 0.67751 0.28992
C 0.52984 0.33851 0.70744
C 0.49952 0.58873 0.18930
C 0.50969 0.42683 0.80774
C 0.44119 0.70211 0.10247
C 0.56754 0.31328 0.89450
C 0.46244 0.61479 0.00401
C 0.54566 0.40036 0.99276
C 0.60346 0.28649 0.08070
C 0.40462 0.72855 0.91603
C 0.58329 0.37502 0.17822
C 0.42538 0.64025 0.81864
C 0.64535 0.26320 0.26882
C 0.36395 0.75226 0.72810
C 0.62625 0.35570 0.36433
C 0.38438 0.66031 0.63293
H 0.18087 0.09674 0.47939
H 0.83091 0.91930 0.52577
H 0.23230 0.31591 0.35792
H 0.77957 0.70019 0.64736
H 0.19550 0.35616 0.17349
H 0.81669 0.65998 0.83178
H 0.85472 0.61973 0.01674
H 0.15727 0.39652 0.98841
H 0.89176 0.58011 0.20170
H 0.11956 0.43633 0.80328
H 0.00921 0.72845 0.40068
H 0.00207 0.28783 0.60435
H 0.52834 0.63557 0.45136
H 0.48368 0.38117 0.54635
H 0.41572 0.83671 0.29767
H 0.59506 0.17954 0.69986
H 0.37794 0.86239 0.11090
H 0.63107 0.15315 0.88618
H 0.66703 0.12635 0.07244
H 0.34097 0.88863 0.92421
H 0.70945 0.10359 0.26056
H 0.29947 0.91163 0.73619
H 0.67493 0.26922 0.43301
H 0.33634 0.74695 0.56430
end atoms_frac
begin projections
random
end projections
begin unit_cell_cart
7.980000 0.000000 0.000000
0.267823 6.134156 0.000000
-5.345791 -2.428121 12.234036
end unit_cell_cart
mp_grid : 6 8 6
begin kpoints
0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.16666667
0.00000000 0.00000000 0.33333333
0.00000000 0.00000000 0.50000000
0.00000000 0.00000000 0.66666667
0.00000000 0.00000000 0.83333333
0.00000000 0.12500000 0.00000000
0.00000000 0.12500000 0.16666667
0.00000000 0.12500000 0.33333333
0.00000000 0.12500000 0.50000000
0.00000000 0.12500000 0.66666667
0.00000000 0.12500000 0.83333333
0.00000000 0.25000000 0.00000000
0.00000000 0.25000000 0.16666667
0.00000000 0.25000000 0.33333333
0.00000000 0.25000000 0.50000000
0.00000000 0.25000000 0.66666667
0.00000000 0.25000000 0.83333333
0.00000000 0.37500000 0.00000000
0.00000000 0.37500000 0.16666667
0.00000000 0.37500000 0.33333333
0.00000000 0.37500000 0.50000000
0.00000000 0.37500000 0.66666667
0.00000000 0.37500000 0.83333333
0.00000000 0.50000000 0.00000000
0.00000000 0.50000000 0.16666667
0.00000000 0.50000000 0.33333333
0.00000000 0.50000000 0.50000000
0.00000000 0.50000000 0.66666667
0.00000000 0.50000000 0.83333333
0.00000000 0.62500000 0.00000000
0.00000000 0.62500000 0.16666667
0.00000000 0.62500000 0.33333333
0.00000000 0.62500000 0.50000000
0.00000000 0.62500000 0.66666667
0.00000000 0.62500000 0.83333333
..............................
--------------------------------------------------------------------
Sincerely,
Jisheng
I don't really know what is initial projections, i have also met some problems.when i plot pentacene's MLWFs using Gaussian cube format, it should be localized on the melecular stucture cause i choose VB and VB-1 bands to construct MLWFs, so it is logically should be like homo and homo-1 in
Gaussian's result. However, my result is not like that. Here is an image of MLWFs: [img][https://i.loli.net/2021/05/18/ItgQnyX4zaVqukC.png]
{kind=link}
Here is my band structure:[img][https://i.loli.net/2021/05/18/eJUycStPOqZo8m1.png], i choose two bands just marked in image. And i've tried wannier_plot_mode = molecule.But it is not good.
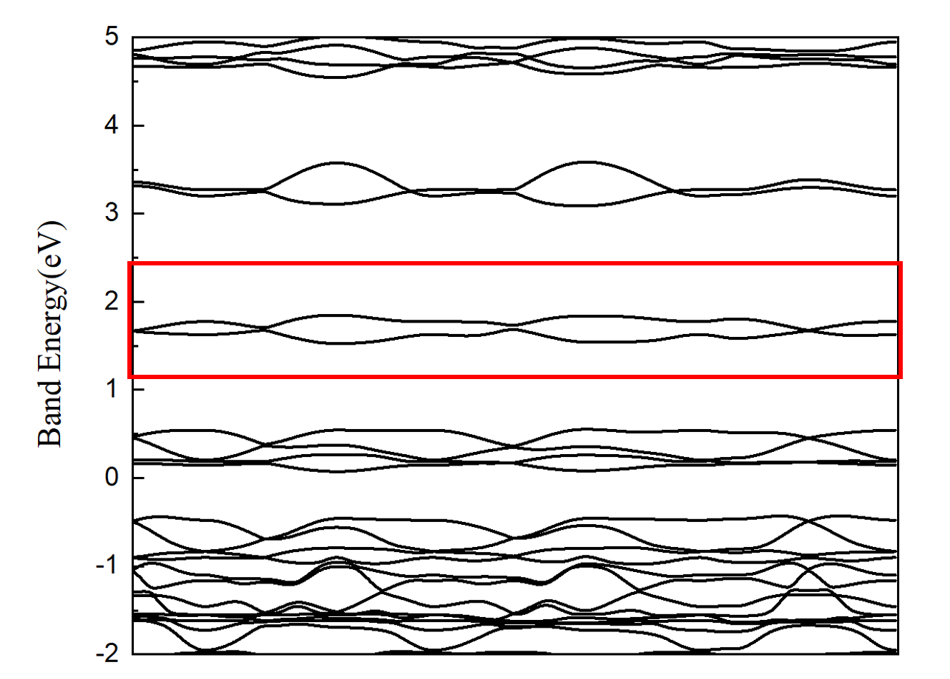{kind=link}
Here is the pdos: [img][https://i.loli.net/2021/05/18/i4YQ8jdqgRDoNXs.png], it is known that C's py orbital is the main contribution to VB and VB-1.
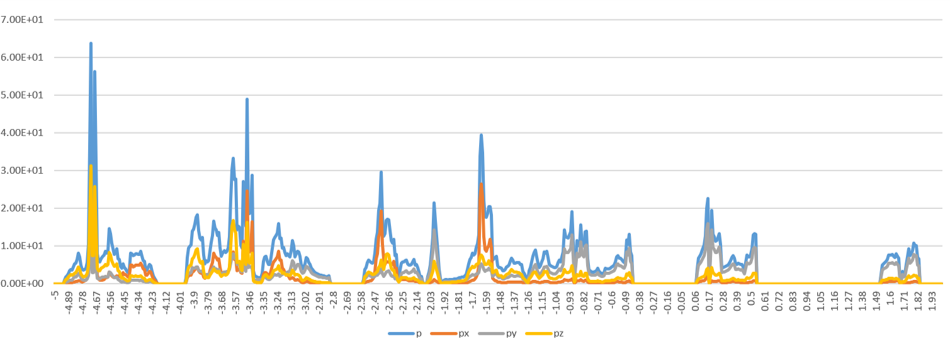{kind=link}
And here are my .win file:
---------------------------------------
exclude_bands = 1:82,85:150
num_wann = 2
num_bands = 2
num_iter = 500
!dis_win_max = 2
!dis_win_min = 2
!dis_froz_max = 1
!dis_froz_min = 1
!dis_num_iter = 50
!dis_mix_ratio = 1.0
kmesh_tol = 0.00001
write_hr= .true.
write_hr_diag=.true.
wannier_plot= .true.
wannier_plot_format = cube
wannier_plot_mode = crystal
wannier_plot_radius = 5
wannier_plot_scale = 1.0
wannier_plot_supercell = 3
wannier_plot_list = 1-2
!bands_plot =.true.
restart = plot
write_xyz=.TRUE.
!gamma_only=.TRUE.
begin atoms_frac
C 0.12959 0.03292 0.39138
C 0.88207 0.98320 0.61377
C 0.15861 0.15508 0.32454
C 0.85321 0.86107 0.68068
C 0.09267 0.07521 0.20891
C 1.91916 0.94108 0.79630
C 0.12093 0.19586 0.13924
C 0.89109 0.82037 0.86600
C 0.05548 0.11521 0.02537
C 0.95656 0.90105 0.97985
C 0.92886 0.78029 0.05088
C 0.08311 0.23596 0.95430
C 0.99391 0.86145 0.16347
C 0.01777 0.15492 0.84169
C 0.96630 0.74018 0.23612
C 0.04512 0.27619 0.76894
C 0.03194 0.82313 0.34656
C 0.97947 0.19315 0.65851
C 0.54261 0.56482 0.37485
C 0.46850 0.45148 0.62262
C 0.48052 0.67751 0.28992
C 0.52984 0.33851 0.70744
C 0.49952 0.58873 0.18930
C 0.50969 0.42683 0.80774
C 0.44119 0.70211 0.10247
C 0.56754 0.31328 0.89450
C 0.46244 0.61479 0.00401
C 0.54566 0.40036 0.99276
C 0.60346 0.28649 0.08070
C 0.40462 0.72855 0.91603
C 0.58329 0.37502 0.17822
C 0.42538 0.64025 0.81864
C 0.64535 0.26320 0.26882
C 0.36395 0.75226 0.72810
C 0.62625 0.35570 0.36433
C 0.38438 0.66031 0.63293
H 0.18087 0.09674 0.47939
H 0.83091 0.91930 0.52577
H 0.23230 0.31591 0.35792
H 0.77957 0.70019 0.64736
H 0.19550 0.35616 0.17349
H 0.81669 0.65998 0.83178
H 0.85472 0.61973 0.01674
H 0.15727 0.39652 0.98841
H 0.89176 0.58011 0.20170
H 0.11956 0.43633 0.80328
H 0.00921 0.72845 0.40068
H 0.00207 0.28783 0.60435
H 0.52834 0.63557 0.45136
H 0.48368 0.38117 0.54635
H 0.41572 0.83671 0.29767
H 0.59506 0.17954 0.69986
H 0.37794 0.86239 0.11090
H 0.63107 0.15315 0.88618
H 0.66703 0.12635 0.07244
H 0.34097 0.88863 0.92421
H 0.70945 0.10359 0.26056
H 0.29947 0.91163 0.73619
H 0.67493 0.26922 0.43301
H 0.33634 0.74695 0.56430
end atoms_frac
begin projections
random
end projections
begin unit_cell_cart
7.980000 0.000000 0.000000
0.267823 6.134156 0.000000
-5.345791 -2.428121 12.234036
end unit_cell_cart
mp_grid : 6 8 6
begin kpoints
0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.16666667
0.00000000 0.00000000 0.33333333
0.00000000 0.00000000 0.50000000
0.00000000 0.00000000 0.66666667
0.00000000 0.00000000 0.83333333
0.00000000 0.12500000 0.00000000
0.00000000 0.12500000 0.16666667
0.00000000 0.12500000 0.33333333
0.00000000 0.12500000 0.50000000
0.00000000 0.12500000 0.66666667
0.00000000 0.12500000 0.83333333
0.00000000 0.25000000 0.00000000
0.00000000 0.25000000 0.16666667
0.00000000 0.25000000 0.33333333
0.00000000 0.25000000 0.50000000
0.00000000 0.25000000 0.66666667
0.00000000 0.25000000 0.83333333
0.00000000 0.37500000 0.00000000
0.00000000 0.37500000 0.16666667
0.00000000 0.37500000 0.33333333
0.00000000 0.37500000 0.50000000
0.00000000 0.37500000 0.66666667
0.00000000 0.37500000 0.83333333
0.00000000 0.50000000 0.00000000
0.00000000 0.50000000 0.16666667
0.00000000 0.50000000 0.33333333
0.00000000 0.50000000 0.50000000
0.00000000 0.50000000 0.66666667
0.00000000 0.50000000 0.83333333
0.00000000 0.62500000 0.00000000
0.00000000 0.62500000 0.16666667
0.00000000 0.62500000 0.33333333
0.00000000 0.62500000 0.50000000
0.00000000 0.62500000 0.66666667
0.00000000 0.62500000 0.83333333
..............................
--------------------------------------------------------------------
Sincerely,
Jisheng
Re: About the projections
Hi Jisheng,
Unfortunately none of the [img] link is working for me. You can attach your image through the attachment. In addition to this, could you share your scf.in, nscf.in, epw.in and band.dat (band structure file which you got from qe).
Best,
Hari
Unfortunately none of the [img] link is working for me. You can attach your image through the attachment. In addition to this, could you share your scf.in, nscf.in, epw.in and band.dat (band structure file which you got from qe).
Best,
Hari
Re: About the projections
Hi Hari!
Thanks for replying. I've made a mistake,the crystal i mentioned above should be tetracene rather than pentacene.
scf.in
----------------------------
&CONTROL
calculation='scf',
restart_mode='from_scratch',
outdir='.',
prefix='tetra',
pseudo_dir='/data/home/jspeng/espresso/pseudo',
forc_conv_thr =1.0D-10
etot_conv_thr = 1.0D-10
verbosity='high'
nstep =500
/
&SYSTEM
ibrav=0,
celldm(1)=15.0800145101d0,
nat=60,
ntyp=2,
occupations = 'smearing'
smearing = 'mp'
degauss = 0.02
nbnd = 150
vdw_corr = 'DFT-D'
ecutwfc=55,
/
&ELECTRONS
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
&IONS
ion_dynamics = 'bfgs',
/
&CELL
cell_dynamics= 'bfgs',
/
ATOMIC_SPECIES
C 12.010700d0 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.129590647 0.032924239 0.391382088
C 0.882065890 0.983201660 0.613769710
C 0.158608098 0.155076409 0.324536811
C 0.853209081 0.861065914 0.680677067
C 0.092672048 0.075210521 0.208905360
C 0.919157497 0.941077084 0.796298973
C 0.120934212 0.195861479 0.139236490
C 0.891093914 0.820374293 0.866001881
C 0.055484347 0.115212764 0.025374413
C 0.956560310 0.901052758 0.979851348
C 0.928861444 0.780293818 0.050883897
C 0.083106748 0.235960362 0.954301440
C 0.993914538 0.861446222 0.163469762
C 0.017773832 0.154916963 0.841686124
C 0.966298289 0.740177602 0.236117543
C 0.045118482 0.276193904 0.768943448
C 0.031935364 0.823128495 0.346555979
C 0.979466046 0.193154883 0.658514471
C 0.542609745 0.564818079 0.374850299
C 0.468495545 0.451480511 0.622624787
C 0.480522821 0.677507122 0.289920802
C 0.529844865 0.338505082 0.707437167
C 0.499516603 0.588729237 0.189302989
C 0.509689637 0.426834329 0.807744691
C 0.441188636 0.702105638 0.102467812
C 0.567540661 0.313280471 0.894497485
C 0.462441677 0.614790804 0.004014496
C 0.545659105 0.400357857 0.992760825
C 0.603460458 0.286486627 0.080697349
C 0.404624843 0.728554375 0.916025092
C 0.583293771 0.375020009 0.178221653
C 0.425381100 0.640250041 0.818636739
C 0.645352871 0.263199234 0.268822610
C 0.363952034 0.752260411 0.728101784
C 0.626247480 0.355701464 0.364331808
C 0.384382244 0.660314461 0.632926730
H 0.180872251 0.096740582 0.479386477
H 0.830912688 0.919300820 0.525772272
H 0.232299332 0.315909316 0.357919323
H 0.779570680 0.700191382 0.647356377
H 0.195501475 0.356159143 0.173494586
H 0.816689155 0.659975943 0.831776602
H 0.854720094 0.619725118 0.016739176
H 0.157270157 0.396517765 0.988412815
H 0.891763480 0.580110790 0.201699210
H 0.119559737 0.436332512 0.803280099
H 0.009209876 0.728445147 0.400675739
H 0.002074339 0.287833428 0.604350332
H 0.528343910 0.635568471 0.451357550
H 0.483675174 0.381166645 0.546347318
H 0.415719685 0.836711796 0.297669453
H 0.595063293 0.179542733 0.699859493
H 0.377944650 0.862392155 0.110895779
H 0.631070697 0.153151209 0.886182637
H 0.667027738 0.126353427 0.072435624
H 0.340965793 0.888634329 0.924213294
H 0.709449712 0.103593623 0.260563380
H 0.299470465 0.911631878 0.736194969
H 0.674925366 0.269222895 0.433014008
H 0.336335560 0.746948278 0.564301262
K_POINTS automatic
3 4 3 0 0 0
CELL_PARAMETERS {alat}
1.000000000000d0 0.000000000000d0 0.000000000000d0
0.033561783454d0 0.768691219601d0 0.000000000000d0
-0.669898611003d0 -0.304275780475d0 1.533087234438d0
-------------------------------------------------------------------------------------------
nscf.in
-------------------------------------------------------------------------
&control
calculation = 'nscf'
prefix = 'tetra'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
forc_conv_thr = 1.0D-8
etot_conv_thr = 1.0D-8
nstep= 500
/
&system
ibrav=0,
celldm(1)=15.0800145101d0,
nat=60,
ntyp=2,
occupations = 'smearing'
smearing = 'mp'
degauss = 0.02
nbnd = 150
vdw_corr = 'DFT-D'
ecutwfc=55,
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
&IONS
ion_dynamics = 'bfgs',
/
&CELL
cell_dynamics= 'bfgs',
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.129590647 0.032924239 0.391382088
C 0.882065890 0.983201660 0.613769710
C 0.158608098 0.155076409 0.324536811
C 0.853209081 0.861065914 0.680677067
C 0.092672048 0.075210521 0.208905360
C 0.919157497 0.941077084 0.796298973
C 0.120934212 0.195861479 0.139236490
C 0.891093914 0.820374293 0.866001881
C 0.055484347 0.115212764 0.025374413
C 0.956560310 0.901052758 0.979851348
C 0.928861444 0.780293818 0.050883897
C 0.083106748 0.235960362 0.954301440
C 0.993914538 0.861446222 0.163469762
C 0.017773832 0.154916963 0.841686124
C 0.966298289 0.740177602 0.236117543
C 0.045118482 0.276193904 0.768943448
C 0.031935364 0.823128495 0.346555979
C 0.979466046 0.193154883 0.658514471
C 0.542609745 0.564818079 0.374850299
C 0.468495545 0.451480511 0.622624787
C 0.480522821 0.677507122 0.289920802
C 0.529844865 0.338505082 0.707437167
C 0.499516603 0.588729237 0.189302989
C 0.509689637 0.426834329 0.807744691
C 0.441188636 0.702105638 0.102467812
C 0.567540661 0.313280471 0.894497485
C 0.462441677 0.614790804 0.004014496
C 0.545659105 0.400357857 0.992760825
C 0.603460458 0.286486627 0.080697349
C 0.404624843 0.728554375 0.916025092
C 0.583293771 0.375020009 0.178221653
C 0.425381100 0.640250041 0.818636739
C 0.645352871 0.263199234 0.268822610
C 0.363952034 0.752260411 0.728101784
C 0.626247480 0.355701464 0.364331808
C 0.384382244 0.660314461 0.632926730
H 0.180872251 0.096740582 0.479386477
H 0.830912688 0.919300820 0.525772272
H 0.232299332 0.315909316 0.357919323
H 0.779570680 0.700191382 0.647356377
H 0.195501475 0.356159143 0.173494586
H 0.816689155 0.659975943 0.831776602
H 0.854720094 0.619725118 0.016739176
H 0.157270157 0.396517765 0.988412815
H 0.891763480 0.580110790 0.201699210
H 0.119559737 0.436332512 0.803280099
H 0.009209876 0.728445147 0.400675739
H 0.002074339 0.287833428 0.604350332
H 0.528343910 0.635568471 0.451357550
H 0.483675174 0.381166645 0.546347318
H 0.415719685 0.836711796 0.297669453
H 0.595063293 0.179542733 0.699859493
H 0.377944650 0.862392155 0.110895779
H 0.631070697 0.153151209 0.886182637
H 0.667027738 0.126353427 0.072435624
H 0.340965793 0.888634329 0.924213294
H 0.709449712 0.103593623 0.260563380
H 0.299470465 0.911631878 0.736194969
H 0.674925366 0.269222895 0.433014008
H 0.336335560 0.746948278 0.564301262
CELL_PARAMETERS {alat}
1.000000000000d0 0.000000000000d0 0.000000000000d0
0.033561783454d0 0.768691219601d0 0.000000000000d0
-0.669898611003d0 -0.304275780475d0 1.533087234438d0
K_POINTS crystal
288
0.00000000 0.00000000 0.00000000 3.472222e-03
0.00000000 0.00000000 0.16666667 3.472222e-03
0.00000000 0.00000000 0.33333333 3.472222e-03
0.00000000 0.00000000 0.50000000 3.472222e-03
0.00000000 0.00000000 0.66666667 3.472222e-03
0.00000000 0.00000000 0.83333333 3.472222e-03
0.00000000 0.12500000 0.00000000 3.472222e-03
0.00000000 0.12500000 0.16666667 3.472222e-03
0.00000000 0.12500000 0.33333333 3.472222e-03
0.00000000 0.12500000 0.50000000 3.472222e-03
0.00000000 0.12500000 0.66666667 3.472222e-03
0.00000000 0.12500000 0.83333333 3.472222e-03
0.00000000 0.25000000 0.00000000 3.472222e-03
0.00000000 0.25000000 0.16666667 3.472222e-03
0.00000000 0.25000000 0.33333333 3.472222e-03
0.00000000 0.25000000 0.50000000 3.472222e-03
0.00000000 0.25000000 0.66666667 3.472222e-03
0.00000000 0.25000000 0.83333333 3.472222e-03
0.00000000 0.37500000 0.00000000 3.472222e-03
0.00000000 0.37500000 0.16666667 3.472222e-03
0.00000000 0.37500000 0.33333333 3.472222e-03
0.00000000 0.37500000 0.50000000 3.472222e-03
0.00000000 0.37500000 0.66666667 3.472222e-03
0.00000000 0.37500000 0.83333333 3.472222e-03
0.00000000 0.50000000 0.00000000 3.472222e-03
0.00000000 0.50000000 0.16666667 3.472222e-03
0.00000000 0.50000000 0.33333333 3.472222e-03
0.00000000 0.50000000 0.50000000 3.472222e-03
0.00000000 0.50000000 0.66666667 3.472222e-03
0.00000000 0.50000000 0.83333333 3.472222e-03
0.00000000 0.62500000 0.00000000 3.472222e-03
0.00000000 0.62500000 0.16666667 3.472222e-03
0.00000000 0.62500000 0.33333333 3.472222e-03
0.00000000 0.62500000 0.50000000 3.472222e-03
0.00000000 0.62500000 0.66666667 3.472222e-03
0.00000000 0.62500000 0.83333333 3.472222e-03
0.00000000 0.75000000 0.00000000 3.472222e-03
0.00000000 0.75000000 0.16666667 3.472222e-03
0.00000000 0.75000000 0.33333333 3.472222e-03
0.00000000 0.75000000 0.50000000 3.472222e-03
................................
---------------------------------------------------------------------------
tetra.win
----------------------------------------------------------------------------
exclude_bands = 1:82,85:150
num_wann = 2
num_bands = 2
num_iter = 500
!dis_win_max = 2
!dis_win_min = 2
!dis_froz_max = 1
!dis_froz_min = 1
!dis_num_iter = 50
!dis_mix_ratio = 1.0
kmesh_tol = 0.00001
write_hr= .true.
write_hr_diag=.true.
wannier_plot= .true.
wannier_plot_format = cube
wannier_plot_mode = crystal
wannier_plot_radius = 5
wannier_plot_scale = 1.0
wannier_plot_supercell = 3
wannier_plot_list = 1-2
bands_plot =.true.
restart = plot
write_xyz=.TRUE.
!gamma_only=.TRUE.
begin kpoint_path
A 0.0000 0.5000 0.00000 G 0.000000 0.000000 0.00000
G 0.000000 0.00000 0.00000 B 0.500000 0.00000 0.0000
B 0.50000 0.00000 0.0000 C 0.50000 0.50000 0.0000
C 0.50000 0.50000 0.0000 G 0.0000 0.0000 0.00000
G 0.0000 0.0000 0.00000 D 0.50000 0.0000 0.50000
D 0.50000 0.00000 0.5000 E 0.5000 0.500000 0.5000
E 0.50000 0.500000 0.5000 G 0.00000 0.00000 0.0000
G 0.00000 0.000000 0.0000 F 0.00000 0.00000 0.5000
F 0.00000 0.000000 0.5000 H 0.00000 0.50000 0.5000
H 0.00000 0.5000 0.5000 G 0.00000 0.00000 0.0000
end kpoint_path
begin atoms_frac
C 0.12959 0.03292 0.39138
C 0.88207 0.98320 0.61377
C 0.15861 0.15508 0.32454
C 0.85321 0.86107 0.68068
C 0.09267 0.07521 0.20891
C 0.91916 0.94108 0.79630
C 0.12093 0.19586 0.13924
C 0.89109 0.82037 0.86600
C 0.05548 0.11521 0.02537
C 0.95656 0.90105 0.97985
C 0.92886 0.78029 0.05088
C 0.08311 0.23596 0.95430
C 0.99391 0.86145 0.16347
C 0.01777 0.15492 0.84169
C 0.96630 0.74018 0.23612
C 0.04512 0.27619 0.76894
C 0.03194 0.82313 0.34656
C 0.97947 0.19315 0.65851
C 0.54261 0.56482 0.37485
C 0.46850 0.45148 0.62262
C 0.48052 0.67751 0.28992
C 0.52984 0.33851 0.70744
C 0.49952 0.58873 0.18930
C 0.50969 0.42683 0.80774
C 0.44119 0.70211 0.10247
C 0.56754 0.31328 0.89450
C 0.46244 0.61479 0.00401
C 0.54566 0.40036 0.99276
C 0.60346 0.28649 0.08070
C 0.40462 0.72855 0.91603
C 0.58329 0.37502 0.17822
C 0.42538 0.64025 0.81864
C 0.64535 0.26320 0.26882
C 0.36395 0.75226 0.72810
C 0.62625 0.35570 0.36433
C 0.38438 0.66031 0.63293
H 0.18087 0.09674 0.47939
H 0.83091 0.91930 0.52577
H 0.23230 0.31591 0.35792
H 0.77957 0.70019 0.64736
H 0.19550 0.35616 0.17349
H 0.81669 0.65998 0.83178
H 0.85472 0.61973 0.01674
H 0.15727 0.39652 0.98841
H 0.89176 0.58011 0.20170
H 0.11956 0.43633 0.80328
H 0.00921 0.72845 0.40068
H 0.00207 0.28783 0.60435
H 0.52834 0.63557 0.45136
H 0.48368 0.38117 0.54635
H 0.41572 0.83671 0.29767
H 0.59506 0.17954 0.69986
H 0.37794 0.86239 0.11090
H 0.63107 0.15315 0.88618
H 0.66703 0.12635 0.07244
H 0.34097 0.88863 0.92421
H 0.70945 0.10359 0.26056
H 0.29947 0.91163 0.73619
H 0.67493 0.26922 0.43301
H 0.33634 0.74695 0.56430
end atoms_frac
begin projections
random
end projections
begin unit_cell_cart
7.980000 0.000000 0.000000
0.267823 6.134156 0.000000
-5.345791 -2.428121 12.234036
end unit_cell_cart
mp_grid : 6 8 6
begin kpoints
0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.16666667
0.00000000 0.00000000 0.33333333
0.00000000 0.00000000 0.50000000
0.00000000 0.00000000 0.66666667
0.00000000 0.00000000 0.83333333
0.00000000 0.12500000 0.00000000
0.00000000 0.12500000 0.16666667
0.00000000 0.12500000 0.33333333
0.00000000 0.12500000 0.50000000
0.00000000 0.12500000 0.66666667
0.00000000 0.12500000 0.83333333
0.00000000 0.25000000 0.00000000
0.00000000 0.25000000 0.16666667
0.00000000 0.25000000 0.33333333
0.00000000 0.25000000 0.50000000
0.00000000 0.25000000 0.66666667
0.00000000 0.25000000 0.83333333
0.00000000 0.37500000 0.00000000
0.00000000 0.37500000 0.16666667
0.00000000 0.37500000 0.33333333
0.00000000 0.37500000 0.50000000
0.00000000 0.37500000 0.66666667
0.00000000 0.37500000 0.83333333
0.00000000 0.50000000 0.00000000
0.00000000 0.50000000 0.16666667
0.00000000 0.50000000 0.33333333
0.00000000 0.50000000 0.50000000
..............................
end kpoints
--------------------------------------------------------------
Thanks for replying. I've made a mistake,the crystal i mentioned above should be tetracene rather than pentacene.
scf.in
----------------------------
&CONTROL
calculation='scf',
restart_mode='from_scratch',
outdir='.',
prefix='tetra',
pseudo_dir='/data/home/jspeng/espresso/pseudo',
forc_conv_thr =1.0D-10
etot_conv_thr = 1.0D-10
verbosity='high'
nstep =500
/
&SYSTEM
ibrav=0,
celldm(1)=15.0800145101d0,
nat=60,
ntyp=2,
occupations = 'smearing'
smearing = 'mp'
degauss = 0.02
nbnd = 150
vdw_corr = 'DFT-D'
ecutwfc=55,
/
&ELECTRONS
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
&IONS
ion_dynamics = 'bfgs',
/
&CELL
cell_dynamics= 'bfgs',
/
ATOMIC_SPECIES
C 12.010700d0 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.129590647 0.032924239 0.391382088
C 0.882065890 0.983201660 0.613769710
C 0.158608098 0.155076409 0.324536811
C 0.853209081 0.861065914 0.680677067
C 0.092672048 0.075210521 0.208905360
C 0.919157497 0.941077084 0.796298973
C 0.120934212 0.195861479 0.139236490
C 0.891093914 0.820374293 0.866001881
C 0.055484347 0.115212764 0.025374413
C 0.956560310 0.901052758 0.979851348
C 0.928861444 0.780293818 0.050883897
C 0.083106748 0.235960362 0.954301440
C 0.993914538 0.861446222 0.163469762
C 0.017773832 0.154916963 0.841686124
C 0.966298289 0.740177602 0.236117543
C 0.045118482 0.276193904 0.768943448
C 0.031935364 0.823128495 0.346555979
C 0.979466046 0.193154883 0.658514471
C 0.542609745 0.564818079 0.374850299
C 0.468495545 0.451480511 0.622624787
C 0.480522821 0.677507122 0.289920802
C 0.529844865 0.338505082 0.707437167
C 0.499516603 0.588729237 0.189302989
C 0.509689637 0.426834329 0.807744691
C 0.441188636 0.702105638 0.102467812
C 0.567540661 0.313280471 0.894497485
C 0.462441677 0.614790804 0.004014496
C 0.545659105 0.400357857 0.992760825
C 0.603460458 0.286486627 0.080697349
C 0.404624843 0.728554375 0.916025092
C 0.583293771 0.375020009 0.178221653
C 0.425381100 0.640250041 0.818636739
C 0.645352871 0.263199234 0.268822610
C 0.363952034 0.752260411 0.728101784
C 0.626247480 0.355701464 0.364331808
C 0.384382244 0.660314461 0.632926730
H 0.180872251 0.096740582 0.479386477
H 0.830912688 0.919300820 0.525772272
H 0.232299332 0.315909316 0.357919323
H 0.779570680 0.700191382 0.647356377
H 0.195501475 0.356159143 0.173494586
H 0.816689155 0.659975943 0.831776602
H 0.854720094 0.619725118 0.016739176
H 0.157270157 0.396517765 0.988412815
H 0.891763480 0.580110790 0.201699210
H 0.119559737 0.436332512 0.803280099
H 0.009209876 0.728445147 0.400675739
H 0.002074339 0.287833428 0.604350332
H 0.528343910 0.635568471 0.451357550
H 0.483675174 0.381166645 0.546347318
H 0.415719685 0.836711796 0.297669453
H 0.595063293 0.179542733 0.699859493
H 0.377944650 0.862392155 0.110895779
H 0.631070697 0.153151209 0.886182637
H 0.667027738 0.126353427 0.072435624
H 0.340965793 0.888634329 0.924213294
H 0.709449712 0.103593623 0.260563380
H 0.299470465 0.911631878 0.736194969
H 0.674925366 0.269222895 0.433014008
H 0.336335560 0.746948278 0.564301262
K_POINTS automatic
3 4 3 0 0 0
CELL_PARAMETERS {alat}
1.000000000000d0 0.000000000000d0 0.000000000000d0
0.033561783454d0 0.768691219601d0 0.000000000000d0
-0.669898611003d0 -0.304275780475d0 1.533087234438d0
-------------------------------------------------------------------------------------------
nscf.in
-------------------------------------------------------------------------
&control
calculation = 'nscf'
prefix = 'tetra'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
forc_conv_thr = 1.0D-8
etot_conv_thr = 1.0D-8
nstep= 500
/
&system
ibrav=0,
celldm(1)=15.0800145101d0,
nat=60,
ntyp=2,
occupations = 'smearing'
smearing = 'mp'
degauss = 0.02
nbnd = 150
vdw_corr = 'DFT-D'
ecutwfc=55,
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
&IONS
ion_dynamics = 'bfgs',
/
&CELL
cell_dynamics= 'bfgs',
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.129590647 0.032924239 0.391382088
C 0.882065890 0.983201660 0.613769710
C 0.158608098 0.155076409 0.324536811
C 0.853209081 0.861065914 0.680677067
C 0.092672048 0.075210521 0.208905360
C 0.919157497 0.941077084 0.796298973
C 0.120934212 0.195861479 0.139236490
C 0.891093914 0.820374293 0.866001881
C 0.055484347 0.115212764 0.025374413
C 0.956560310 0.901052758 0.979851348
C 0.928861444 0.780293818 0.050883897
C 0.083106748 0.235960362 0.954301440
C 0.993914538 0.861446222 0.163469762
C 0.017773832 0.154916963 0.841686124
C 0.966298289 0.740177602 0.236117543
C 0.045118482 0.276193904 0.768943448
C 0.031935364 0.823128495 0.346555979
C 0.979466046 0.193154883 0.658514471
C 0.542609745 0.564818079 0.374850299
C 0.468495545 0.451480511 0.622624787
C 0.480522821 0.677507122 0.289920802
C 0.529844865 0.338505082 0.707437167
C 0.499516603 0.588729237 0.189302989
C 0.509689637 0.426834329 0.807744691
C 0.441188636 0.702105638 0.102467812
C 0.567540661 0.313280471 0.894497485
C 0.462441677 0.614790804 0.004014496
C 0.545659105 0.400357857 0.992760825
C 0.603460458 0.286486627 0.080697349
C 0.404624843 0.728554375 0.916025092
C 0.583293771 0.375020009 0.178221653
C 0.425381100 0.640250041 0.818636739
C 0.645352871 0.263199234 0.268822610
C 0.363952034 0.752260411 0.728101784
C 0.626247480 0.355701464 0.364331808
C 0.384382244 0.660314461 0.632926730
H 0.180872251 0.096740582 0.479386477
H 0.830912688 0.919300820 0.525772272
H 0.232299332 0.315909316 0.357919323
H 0.779570680 0.700191382 0.647356377
H 0.195501475 0.356159143 0.173494586
H 0.816689155 0.659975943 0.831776602
H 0.854720094 0.619725118 0.016739176
H 0.157270157 0.396517765 0.988412815
H 0.891763480 0.580110790 0.201699210
H 0.119559737 0.436332512 0.803280099
H 0.009209876 0.728445147 0.400675739
H 0.002074339 0.287833428 0.604350332
H 0.528343910 0.635568471 0.451357550
H 0.483675174 0.381166645 0.546347318
H 0.415719685 0.836711796 0.297669453
H 0.595063293 0.179542733 0.699859493
H 0.377944650 0.862392155 0.110895779
H 0.631070697 0.153151209 0.886182637
H 0.667027738 0.126353427 0.072435624
H 0.340965793 0.888634329 0.924213294
H 0.709449712 0.103593623 0.260563380
H 0.299470465 0.911631878 0.736194969
H 0.674925366 0.269222895 0.433014008
H 0.336335560 0.746948278 0.564301262
CELL_PARAMETERS {alat}
1.000000000000d0 0.000000000000d0 0.000000000000d0
0.033561783454d0 0.768691219601d0 0.000000000000d0
-0.669898611003d0 -0.304275780475d0 1.533087234438d0
K_POINTS crystal
288
0.00000000 0.00000000 0.00000000 3.472222e-03
0.00000000 0.00000000 0.16666667 3.472222e-03
0.00000000 0.00000000 0.33333333 3.472222e-03
0.00000000 0.00000000 0.50000000 3.472222e-03
0.00000000 0.00000000 0.66666667 3.472222e-03
0.00000000 0.00000000 0.83333333 3.472222e-03
0.00000000 0.12500000 0.00000000 3.472222e-03
0.00000000 0.12500000 0.16666667 3.472222e-03
0.00000000 0.12500000 0.33333333 3.472222e-03
0.00000000 0.12500000 0.50000000 3.472222e-03
0.00000000 0.12500000 0.66666667 3.472222e-03
0.00000000 0.12500000 0.83333333 3.472222e-03
0.00000000 0.25000000 0.00000000 3.472222e-03
0.00000000 0.25000000 0.16666667 3.472222e-03
0.00000000 0.25000000 0.33333333 3.472222e-03
0.00000000 0.25000000 0.50000000 3.472222e-03
0.00000000 0.25000000 0.66666667 3.472222e-03
0.00000000 0.25000000 0.83333333 3.472222e-03
0.00000000 0.37500000 0.00000000 3.472222e-03
0.00000000 0.37500000 0.16666667 3.472222e-03
0.00000000 0.37500000 0.33333333 3.472222e-03
0.00000000 0.37500000 0.50000000 3.472222e-03
0.00000000 0.37500000 0.66666667 3.472222e-03
0.00000000 0.37500000 0.83333333 3.472222e-03
0.00000000 0.50000000 0.00000000 3.472222e-03
0.00000000 0.50000000 0.16666667 3.472222e-03
0.00000000 0.50000000 0.33333333 3.472222e-03
0.00000000 0.50000000 0.50000000 3.472222e-03
0.00000000 0.50000000 0.66666667 3.472222e-03
0.00000000 0.50000000 0.83333333 3.472222e-03
0.00000000 0.62500000 0.00000000 3.472222e-03
0.00000000 0.62500000 0.16666667 3.472222e-03
0.00000000 0.62500000 0.33333333 3.472222e-03
0.00000000 0.62500000 0.50000000 3.472222e-03
0.00000000 0.62500000 0.66666667 3.472222e-03
0.00000000 0.62500000 0.83333333 3.472222e-03
0.00000000 0.75000000 0.00000000 3.472222e-03
0.00000000 0.75000000 0.16666667 3.472222e-03
0.00000000 0.75000000 0.33333333 3.472222e-03
0.00000000 0.75000000 0.50000000 3.472222e-03
................................
---------------------------------------------------------------------------
tetra.win
----------------------------------------------------------------------------
exclude_bands = 1:82,85:150
num_wann = 2
num_bands = 2
num_iter = 500
!dis_win_max = 2
!dis_win_min = 2
!dis_froz_max = 1
!dis_froz_min = 1
!dis_num_iter = 50
!dis_mix_ratio = 1.0
kmesh_tol = 0.00001
write_hr= .true.
write_hr_diag=.true.
wannier_plot= .true.
wannier_plot_format = cube
wannier_plot_mode = crystal
wannier_plot_radius = 5
wannier_plot_scale = 1.0
wannier_plot_supercell = 3
wannier_plot_list = 1-2
bands_plot =.true.
restart = plot
write_xyz=.TRUE.
!gamma_only=.TRUE.
begin kpoint_path
A 0.0000 0.5000 0.00000 G 0.000000 0.000000 0.00000
G 0.000000 0.00000 0.00000 B 0.500000 0.00000 0.0000
B 0.50000 0.00000 0.0000 C 0.50000 0.50000 0.0000
C 0.50000 0.50000 0.0000 G 0.0000 0.0000 0.00000
G 0.0000 0.0000 0.00000 D 0.50000 0.0000 0.50000
D 0.50000 0.00000 0.5000 E 0.5000 0.500000 0.5000
E 0.50000 0.500000 0.5000 G 0.00000 0.00000 0.0000
G 0.00000 0.000000 0.0000 F 0.00000 0.00000 0.5000
F 0.00000 0.000000 0.5000 H 0.00000 0.50000 0.5000
H 0.00000 0.5000 0.5000 G 0.00000 0.00000 0.0000
end kpoint_path
begin atoms_frac
C 0.12959 0.03292 0.39138
C 0.88207 0.98320 0.61377
C 0.15861 0.15508 0.32454
C 0.85321 0.86107 0.68068
C 0.09267 0.07521 0.20891
C 0.91916 0.94108 0.79630
C 0.12093 0.19586 0.13924
C 0.89109 0.82037 0.86600
C 0.05548 0.11521 0.02537
C 0.95656 0.90105 0.97985
C 0.92886 0.78029 0.05088
C 0.08311 0.23596 0.95430
C 0.99391 0.86145 0.16347
C 0.01777 0.15492 0.84169
C 0.96630 0.74018 0.23612
C 0.04512 0.27619 0.76894
C 0.03194 0.82313 0.34656
C 0.97947 0.19315 0.65851
C 0.54261 0.56482 0.37485
C 0.46850 0.45148 0.62262
C 0.48052 0.67751 0.28992
C 0.52984 0.33851 0.70744
C 0.49952 0.58873 0.18930
C 0.50969 0.42683 0.80774
C 0.44119 0.70211 0.10247
C 0.56754 0.31328 0.89450
C 0.46244 0.61479 0.00401
C 0.54566 0.40036 0.99276
C 0.60346 0.28649 0.08070
C 0.40462 0.72855 0.91603
C 0.58329 0.37502 0.17822
C 0.42538 0.64025 0.81864
C 0.64535 0.26320 0.26882
C 0.36395 0.75226 0.72810
C 0.62625 0.35570 0.36433
C 0.38438 0.66031 0.63293
H 0.18087 0.09674 0.47939
H 0.83091 0.91930 0.52577
H 0.23230 0.31591 0.35792
H 0.77957 0.70019 0.64736
H 0.19550 0.35616 0.17349
H 0.81669 0.65998 0.83178
H 0.85472 0.61973 0.01674
H 0.15727 0.39652 0.98841
H 0.89176 0.58011 0.20170
H 0.11956 0.43633 0.80328
H 0.00921 0.72845 0.40068
H 0.00207 0.28783 0.60435
H 0.52834 0.63557 0.45136
H 0.48368 0.38117 0.54635
H 0.41572 0.83671 0.29767
H 0.59506 0.17954 0.69986
H 0.37794 0.86239 0.11090
H 0.63107 0.15315 0.88618
H 0.66703 0.12635 0.07244
H 0.34097 0.88863 0.92421
H 0.70945 0.10359 0.26056
H 0.29947 0.91163 0.73619
H 0.67493 0.26922 0.43301
H 0.33634 0.74695 0.56430
end atoms_frac
begin projections
random
end projections
begin unit_cell_cart
7.980000 0.000000 0.000000
0.267823 6.134156 0.000000
-5.345791 -2.428121 12.234036
end unit_cell_cart
mp_grid : 6 8 6
begin kpoints
0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.16666667
0.00000000 0.00000000 0.33333333
0.00000000 0.00000000 0.50000000
0.00000000 0.00000000 0.66666667
0.00000000 0.00000000 0.83333333
0.00000000 0.12500000 0.00000000
0.00000000 0.12500000 0.16666667
0.00000000 0.12500000 0.33333333
0.00000000 0.12500000 0.50000000
0.00000000 0.12500000 0.66666667
0.00000000 0.12500000 0.83333333
0.00000000 0.25000000 0.00000000
0.00000000 0.25000000 0.16666667
0.00000000 0.25000000 0.33333333
0.00000000 0.25000000 0.50000000
0.00000000 0.25000000 0.66666667
0.00000000 0.25000000 0.83333333
0.00000000 0.37500000 0.00000000
0.00000000 0.37500000 0.16666667
0.00000000 0.37500000 0.33333333
0.00000000 0.37500000 0.50000000
0.00000000 0.37500000 0.66666667
0.00000000 0.37500000 0.83333333
0.00000000 0.50000000 0.00000000
0.00000000 0.50000000 0.16666667
0.00000000 0.50000000 0.33333333
0.00000000 0.50000000 0.50000000
..............................
end kpoints
--------------------------------------------------------------
- Attachments
-
- 图片3.png (93 KiB) Viewed 14201 times
-
- 图片2.png (86.32 KiB) Viewed 14201 times
-
- 图片1.png (127.08 KiB) Viewed 14201 times
Re: About the projections
Dear Jisheng:
I don't know about Gaussians, but Wannier functions are not the eigenstates of Hamiltonian H(k).
Additionally, as I said, in general, random projections are not good initial projections, but in your specific case in which only a few bands constitute a band manifold of interest, it is non-trivial to choose (or guess) the (good) initial projections and it is worth trying the random projections instead.
Sincerely,
H. Lee
I don't know about Gaussians, but Wannier functions are not the eigenstates of Hamiltonian H(k).
Additionally, as I said, in general, random projections are not good initial projections, but in your specific case in which only a few bands constitute a band manifold of interest, it is non-trivial to choose (or guess) the (good) initial projections and it is worth trying the random projections instead.
Sincerely,
H. Lee