Dear all,
I've calculated naphthalene electron-phonon vertex by qe-6.4.1 and EPW v.5.1.0. But the result is too large.
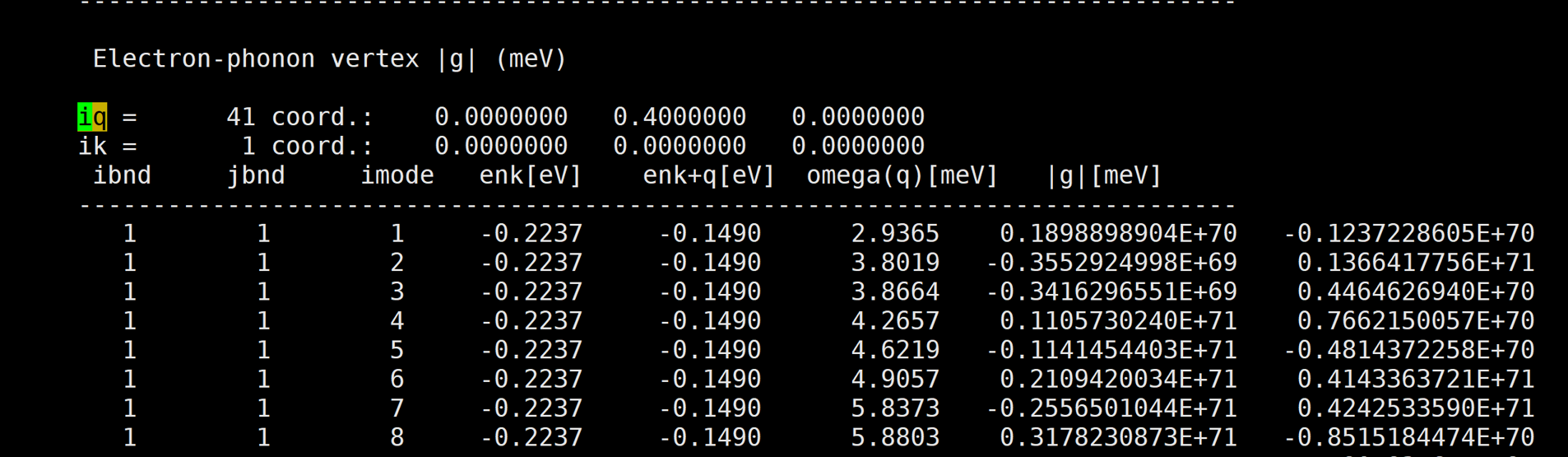
Here is the result: [img][https://i.loli.net/2020/08/19/PYzZAoGXFMHu9mJ.png]
And the input file i use is
scf.in
-------------------------------------------------------------------------------------
&control
calculation = 'scf'
prefix = 'napht'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
/
&system
ibrav=0,
celldm(1)= 15.32199397
nat=36,
ntyp=2,
ecutwfc=60,
ecutrho=600,
occupations = 'smearing'
smearing = 'gauss'
degauss = 1.0d-9
nbnd = 96
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.081425852 0.016578000 0.320425540
C 0.418563591 0.516578633 0.679561956
C 0.918568223 0.983429349 0.679566194
C 0.581425272 0.483420039 0.320421614
C 0.109536973 0.152891341 0.216967674
C 0.390453781 0.652894793 0.783031949
C 0.890450629 0.847099642 0.783026330
C 0.609537634 0.347109257 0.216971160
C 0.046735358 0.099521927 0.036652394
C 0.453254739 0.599524315 0.963339212
C 0.953256278 0.900476146 0.963345888
C 0.546738013 0.400474080 0.036652955
C 0.073281485 0.236543501 0.926610989
C 0.426711824 0.736536528 0.073385646
C 0.926711917 0.763465068 0.073383309
C 0.573273521 0.263470909 0.926599644
C 0.989266875 0.820195453 0.248086166
C 0.510729800 0.320187579 0.751919324
C 0.010721643 0.179799473 0.751907756
C 0.489267085 0.679804682 0.248087034
H 0.130354682 0.060288819 0.458678341
H 0.369637221 0.560287937 0.541319067
H 0.869636610 0.939711193 0.541317391
H 0.630354818 0.439709201 0.458679729
H 0.179868851 0.304754782 0.271710803
H 0.320123069 0.804753111 0.728283974
H 0.820123570 0.695248369 0.728284733
H 0.679868138 0.195245174 0.271710031
H 0.144899815 0.387460949 0.982909846
H 0.355088380 0.887461819 0.017083198
H 0.855088700 0.612536665 0.017083609
H 0.644903165 0.112531931 0.982912660
H 0.967281381 0.713650483 0.330850532
H 0.532708051 0.213649357 0.669142406
H 0.032708263 0.286351359 0.669142611
H 0.467280791 0.786348134 0.330850337
K_POINTS automatic
4 6 4 0 0 0
CELL_PARAMETERS (alat=15.32199397)
1.070650066 -0.000000900 0.015729322
-0.000000687 0.776651697 -0.000000193
-0.631530124 -0.000000272 0.902091712
------------------------------------------------------------------------------------
nscf_epw.in
------------------------------------------------------------------------------------
&control
calculation = 'nscf'
prefix = 'napht'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
/
&system
ibrav=0,
celldm(1)= 15.32199397
nat=36,
ntyp=2,
ecutwfc=60,
ecutrho=600,
occupations = 'smearing'
smearing = 'gauss'
degauss = 1.0d-9
nbnd = 96
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.081425852 0.016578000 0.320425540
C 0.418563591 0.516578633 0.679561956
C 0.918568223 0.983429349 0.679566194
C 0.581425272 0.483420039 0.320421614
C 0.109536973 0.152891341 0.216967674
C 0.390453781 0.652894793 0.783031949
C 0.890450629 0.847099642 0.783026330
.................
............................
..............................................
CELL_PARAMETERS (alat=15.32199397)
1.070650066 -0.000000900 0.015729322
-0.000000687 0.776651697 -0.000000193
-0.631530124 -0.000000272 0.902091712
K_POINTS crystal
96
0.00000000 0.00000000 0.00000000 1.041667e-02
0.00000000 0.00000000 0.25000000 1.041667e-02
0.00000000 0.00000000 0.50000000 1.041667e-02
0.00000000 0.00000000 0.75000000 1.041667e-02
0.00000000 0.16666667 0.00000000 1.041667e-02
0.00000000 0.16666667 0.25000000 1.041667e-02
0.00000000 0.16666667 0.50000000 1.041667e-02
0.00000000 0.16666667 0.75000000 1.041667e-02
0.00000000 0.33333333 0.00000000 1.041667e-02
0.00000000 0.33333333 0.25000000 1.041667e-02
0.00000000 0.33333333 0.50000000 1.041667e-02
..................................
........................................
...............................................
------------------------------------------------------------------------------------------
epw.in
------------------------------------------------------------------------------------------
--
&inputepw
prefix = 'napht'
amass(1) = 12.01078
amass(2) = 1.007940
outdir = './'
dvscf_dir = './save/'
iverbosity = 1
elph = .true.
kmaps = .false.
epbwrite = .true.
epbread = .false.
! etf_mem = 2 !all the fine Bloch-space el-ph matrix elements are store
epwwrite = .true.
epwread = .false.
nbndsub = 2
nbndskip = 46
bands_skipped = 'exclude_bands = 1:46,49:96'
wannierize = .true.
num_iter = 800
iprint = 2
! dis_win_max = 8
! dis_froz_max= 5.5
!proj(1) = 'Si : sp3'
proj(1) = 'random'
! wdata(1) = 'bands_plot= .true.'
wdata(8) = 'kmesh_tol = 0.00001'
! wdata(2) = 'begin kpoint_path'
! wdata(3) = 'Y 0.5 0 0 G 0 0 0'
! wdata(4) = 'G 0 0 0 B 0 0.5 0'
! wdata(5) = 'B 0 0.5 0 G 0 0 0'
! wdata(6) = 'G 0 0 0 Z 0 0 0.5'
! wdata(7) = 'end kpoint_path'
elecselfen = .false.
phonselfen = .false.
a2f = .false.
fsthick = 20 ! eV
eptemp = 1 ! K
degaussw = 0.01 ! eV
nkf1 = 10
nkf2 = 10
nkf3 = 10
nqf1 = 10
nqf2 = 10
nqf3 = 10
nk1 = 4
nk2 = 6
nk3 = 4
nq1 = 2
nq2 = 3
nq3 = 2
/
8 cartesian
0.000000000000000E+00 0.000000000000000E+00 0.000000000000000E+00
0.806005204269855E-02 -0.129205048000064E-06 -0.548624743748963E+00
0.355229347794158E-06 0.429192821725915E+00 0.378097345350993E-06
0.806040727204635E-02 0.429192692520867E+00 -0.548624365651617E+00
-0.462251719820465E+00 -0.489310279249972E-06 -0.323609985608164E+00
-0.454191667777766E+00 -0.618515327250036E-06 -0.872234729357127E+00
-0.462251364591117E+00 0.429192332415636E+00 -0.323609607510819E+00
-0.454191312548419E+00 0.429192203210588E+00 -0.872234351259782E+00
-------------------------------------------------------------------------------------------------------
epw2.in
-------------------------------------------------------------------------------------------------------
--
&inputepw
prefix = 'napht'
amass(1) = 12.01078
amass(2) = 1.007940
outdir = './'
dvscf_dir = './save/'
iverbosity = 3
elph = .true.
kmaps = .true.
epbwrite = .false.
epbread = .true.
etf_mem = 2 !all the fine Bloch-space el-ph matrix elements are store
epwwrite = .false.
epwread = .true.
lifc = .true.
asr_typ = 'simple'
nbndsub = 2
nbndskip = 46
bands_skipped = 'exclude_bands = 1:46,49:96'
wannierize = .false.
num_iter = 800
iprint = 2
! dis_win_max = 8
! dis_froz_max= 5.5
!proj(1) = 'Si : sp3'
proj(1) = 'random'
wdata(1) = 'bands_plot= .true.'
wdata(9) = 'kmesh_tol = 0.0001'
wdata(2) = 'begin kpoint_path'
wdata(3) = 'Y 0.5 0 0 G 0 0 0'
wdata(4) = 'G 0 0 0 B 0 0.5 0'
wdata(5) = 'B 0 0.5 0 G 0 0 0'
wdata(6) = 'G 0 0 0 Z 0 0 0.5'
wdata(7) = 'end kpoint_path'
wdata(8) = 'bands_num_points = 50'
elecselfen = .false.
phonselfen = .false.
a2f = .false.
band_plot = .false.
fsthick = 20 ! eV
eptemp = 1 ! K
degaussw = 0.01 ! eV
! filqf ='napht_band.qpt'
! filkf ='napht_band.kpt'
nkf1 = 10
nkf2 = 10
nkf3 = 10
nqf1 = 10
nqf2 = 10
nqf3 = 10
nk1 = 4
nk2 = 6
nk3 = 4
nq1 = 2
nq2 = 3
nq3 = 2
/
8 cartesian
0.000000000000000E+00 0.000000000000000E+00 0.000000000000000E+00
0.806005204269855E-02 -0.129205048000064E-06 -0.548624743748963E+00
0.355229347794158E-06 0.429192821725915E+00 0.378097345350993E-06
..................
.............................
............................................
----------------------------------------------------------------------------------------------------------
I don't know where did i go wrong, if you have any idea,please tell me! Thanks a lot!
sincerely,
Jisheng
The electron-phonon vertex calculated by EPW is too large
Moderator: stiwari
{kind=link}
Re: The electron-phonon vertex calculated by EPW is too large
Dear Jisheng:
You attached the result for |g| at k=0 and k+q=0.
Did you obtain such large |g| at other k and q pairs as well?
Additionally, as I already mentioned in my previous post about your different question, EPW v5.1 doesn't support PAW; while this might not be the direct source of your issue, EPW v5.1 misses the PAW contribution to the perturbing potential (dvscf files).
(Excerpt from my previous post)
H. Lee
You attached the result for |g| at k=0 and k+q=0.
Did you obtain such large |g| at other k and q pairs as well?
Additionally, as I already mentioned in my previous post about your different question, EPW v5.1 doesn't support PAW; while this might not be the direct source of your issue, EPW v5.1 misses the PAW contribution to the perturbing potential (dvscf files).
(Excerpt from my previous post)
Sincerely,First, EPW v5.1 doesn't support PAW while I think that it is not directly related to your issue as far as the phonon dispersion is concerned.
I would suggest you to try the recent EPW v5.3 included in QE v6.6 at https://gitlab.com/QEF/q-e/-/releases .
Compared with EPW v5.1, there are several changes in EPW v5.3; please read carefully the Release notes at https://docs.epw-code.org/doc/Releases.html#epw-v5-3 .
You might need to perform calculations from scratch.
H. Lee
Re: The electron-phonon vertex calculated by EPW is too large
Dear H. Lee,
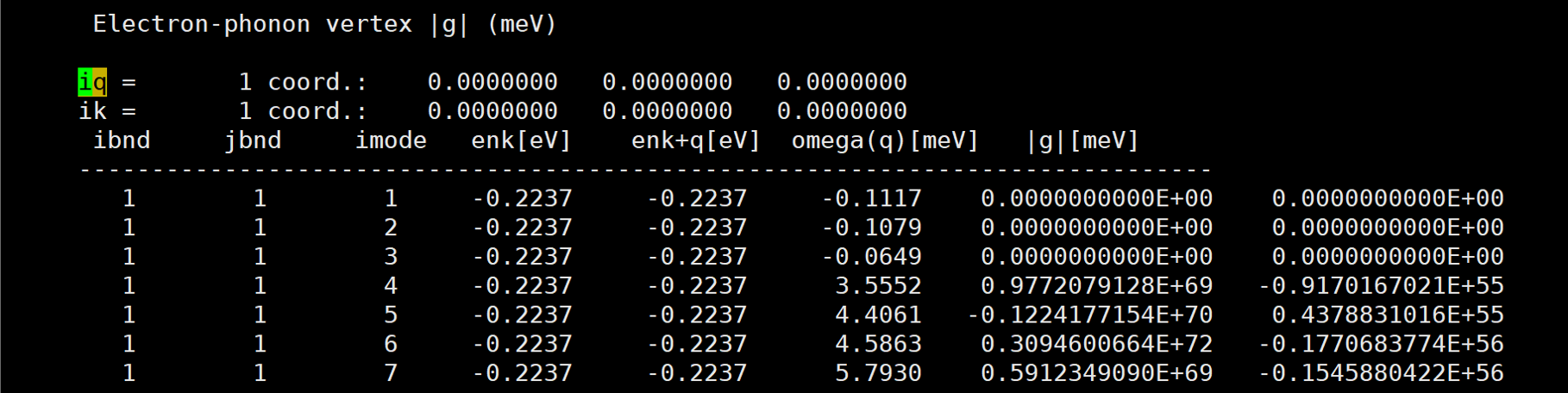
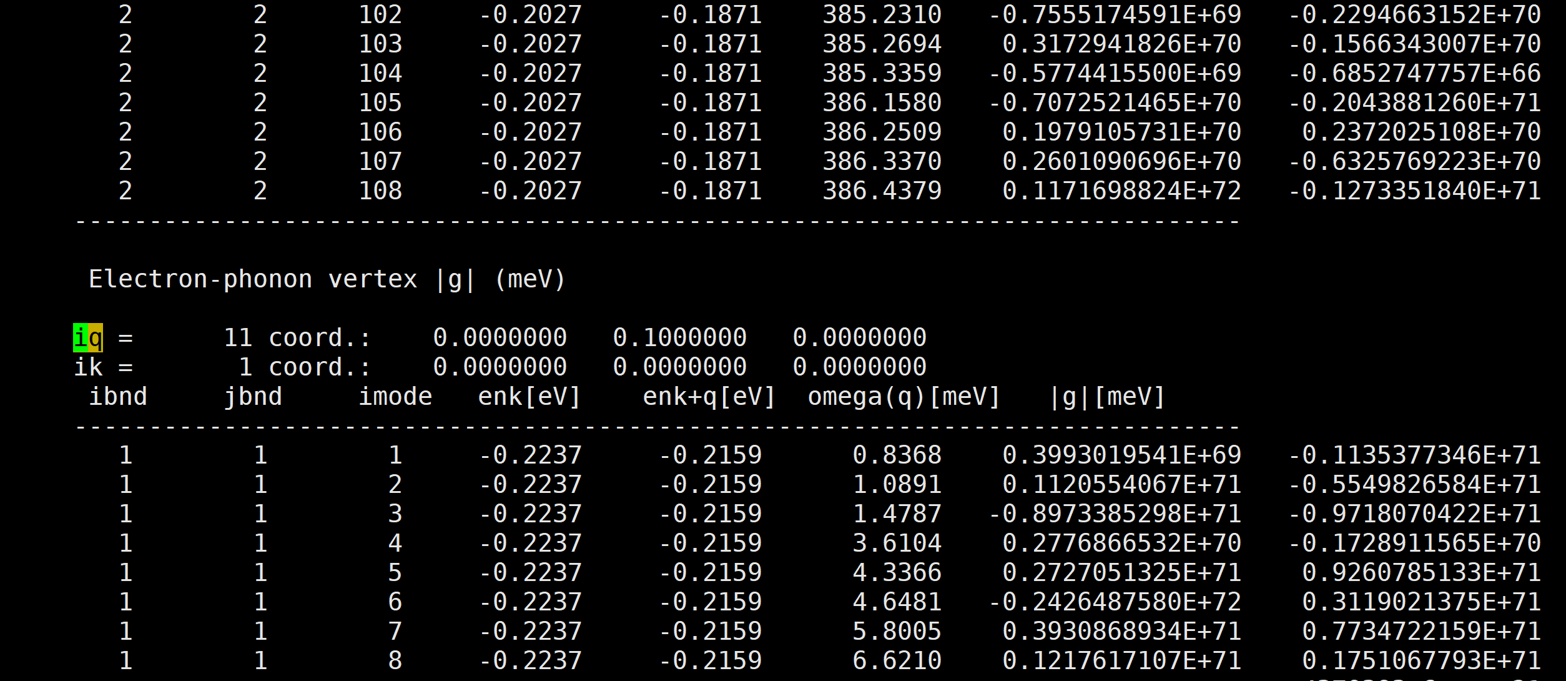
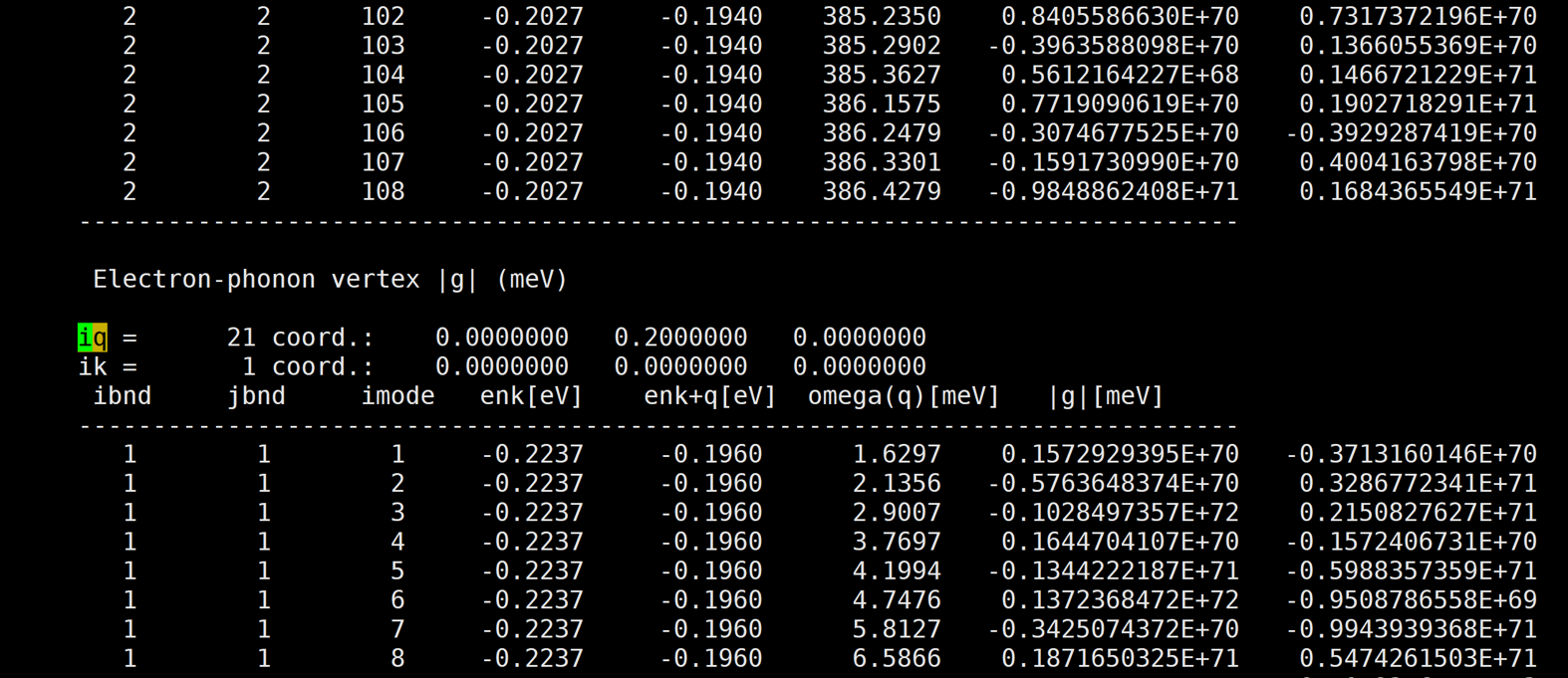
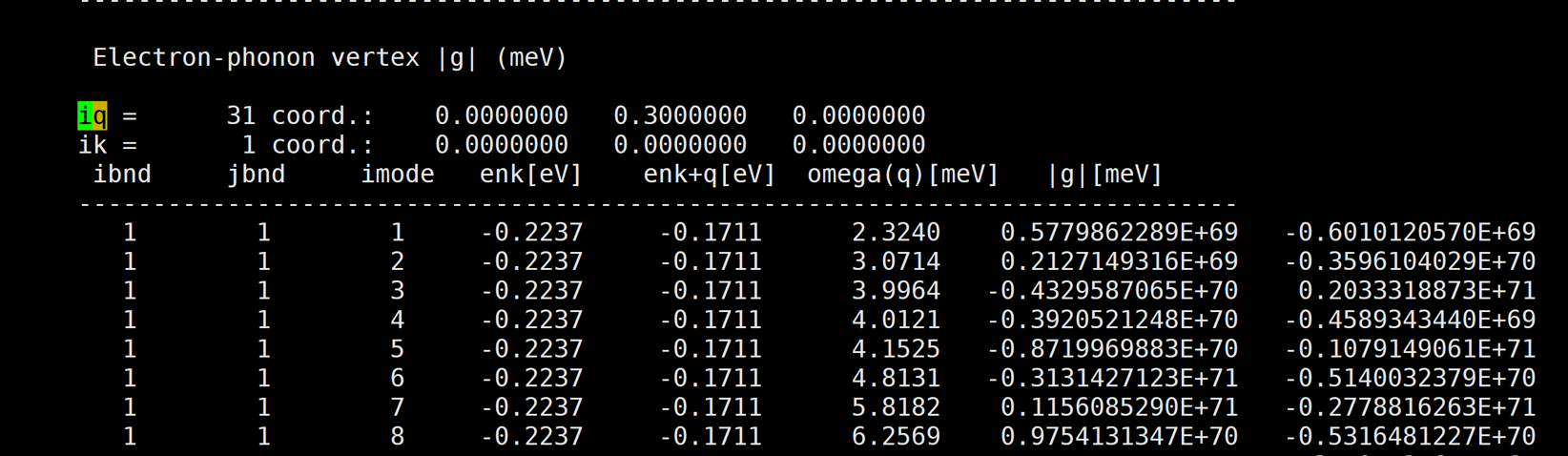
Glad to see you again! Here are more results at other k and q pairs.
[img][https://i.loli.net/2020/08/20/xUos8y9et1DWKIT.png]
[img][https://i.loli.net/2020/08/20/nRXacvBmlSj8dU9.png]
[img][https://i.loli.net/2020/08/20/bdErXa5GTKQPUAe.png]
[img][https://i.loli.net/2020/08/20/WsvUOPrKx36QAZf.png]
And almost at every k and q pair, the results are very large.
The first number of |g| is the real part, and the second number of |g| is the imaginary part.
A member of my group also use qe-6.4.1 and EPW v5.1 to calculate naphthalene's |g|, but her's results are very good. However, our inputs are almost the same except the atom position. So i am very confused.
Sincerely,
Jisheng
Glad to see you again! Here are more results at other k and q pairs.
[img][https://i.loli.net/2020/08/20/xUos8y9et1DWKIT.png]
{kind=link}
[img][https://i.loli.net/2020/08/20/nRXacvBmlSj8dU9.png]
{kind=link}
[img][https://i.loli.net/2020/08/20/bdErXa5GTKQPUAe.png]
{kind=link}
[img][https://i.loli.net/2020/08/20/WsvUOPrKx36QAZf.png]
{kind=link}
And almost at every k and q pair, the results are very large.
The first number of |g| is the real part, and the second number of |g| is the imaginary part.
A member of my group also use qe-6.4.1 and EPW v5.1 to calculate naphthalene's |g|, but her's results are very good. However, our inputs are almost the same except the atom position. So i am very confused.
Sincerely,
Jisheng
Re: The electron-phonon vertex calculated by EPW is too large
Dear Jisheng:
As I said several times, in principle, you should not use EPW v5.1 with PAW while the PAW correction to the perturbing potential and dynamics matrices, etc. might be small.
Also if your colleague's results are different from yours, please compare VERY carefully all inputs and outputs (bands, phonon dispersions, wannier functions, etc.) (I assume that your code is the exact same version as your colleague's code) to identify where the difference occurs.
Sincerely,
H. Lee
As I said several times, in principle, you should not use EPW v5.1 with PAW while the PAW correction to the perturbing potential and dynamics matrices, etc. might be small.
Also if your colleague's results are different from yours, please compare VERY carefully all inputs and outputs (bands, phonon dispersions, wannier functions, etc.) (I assume that your code is the exact same version as your colleague's code) to identify where the difference occurs.
Sincerely,
H. Lee
Re: The electron-phonon vertex calculated by EPW is too large
Dear H. Lee,
After following your advice, i compare very carefully with my colleague's input ,finally i've found my mistake, i used dagauss= 10E-9 when i ran scf.in. As i changed degauss to 0.02(same as my colleague's input), the magnitude of the calculated electron-phonon vertex became reasonable. But here comes another question. What is the criteria to choose a proper degauss? What value of degauss is suitable for metal and what is for semiconductor? And what is the reason? Thanks a lot!
Sincerely,
Jisheng
After following your advice, i compare very carefully with my colleague's input ,finally i've found my mistake, i used dagauss= 10E-9 when i ran scf.in. As i changed degauss to 0.02(same as my colleague's input), the magnitude of the calculated electron-phonon vertex became reasonable. But here comes another question. What is the criteria to choose a proper degauss? What value of degauss is suitable for metal and what is for semiconductor? And what is the reason? Thanks a lot!
Sincerely,
Jisheng
Re: The electron-phonon vertex calculated by EPW is too large
Dear Jisheng:
degauss is relevant for metals and narrow-gap semiconductors.
It is one of the convergence parameters in DFT calculations and it is closely related to the number of k points.
If you want to achieve the convergence with very small degauss as in your inputs (dagauss= 10E-9), you really need very large number of k points.
For details of degauss, please check any introductory DFT book (for instance, https://www.amazon.com/Density-Function ... 0470373172 ) or the webpages on the DFT calculations. For the latter, when googling you can use the keywords of "smearing" and "degauss", etc.
PS) As I said several times, in principle, you should not use EPW v5.1 with PAW while the PAW correction to the perturbing potential and dynamics matrices, etc. might be small.
Sincerely,
H. Lee
degauss is relevant for metals and narrow-gap semiconductors.
It is one of the convergence parameters in DFT calculations and it is closely related to the number of k points.
If you want to achieve the convergence with very small degauss as in your inputs (dagauss= 10E-9), you really need very large number of k points.
For details of degauss, please check any introductory DFT book (for instance, https://www.amazon.com/Density-Function ... 0470373172 ) or the webpages on the DFT calculations. For the latter, when googling you can use the keywords of "smearing" and "degauss", etc.
PS) As I said several times, in principle, you should not use EPW v5.1 with PAW while the PAW correction to the perturbing potential and dynamics matrices, etc. might be small.
Sincerely,
H. Lee
Re: The electron-phonon vertex calculated by EPW is too large
Dear H. Lee,
Thanks a lot for your reply, i'll take the newest version of QE and EPW when i finally choose the system i need to calculate. Naphthalene is just for practice, so i need advices from my colleagues and guys like you. It's lucky to have advisor in this forum!
Sincerely,
Jisheng
Thanks a lot for your reply, i'll take the newest version of QE and EPW when i finally choose the system i need to calculate. Naphthalene is just for practice, so i need advices from my colleagues and guys like you. It's lucky to have advisor in this forum!
Sincerely,
Jisheng
Re: The electron-phonon vertex calculated by EPW is too large
Dear Jisheng:
I think that Naphthalene is too complex a material for practice.
If you have not yet checked, please check the page of the 2018 School on Electron-Phonon Physics from First Principles at https://docs.epw-code.org/doc/School2018.html and the tutorials therein.
Additionally, you can check the Tutorial page at https://docs.epw-code.org/doc/Tutorial.html .
Sincerely,
H. Lee
I think that Naphthalene is too complex a material for practice.
If you have not yet checked, please check the page of the 2018 School on Electron-Phonon Physics from First Principles at https://docs.epw-code.org/doc/School2018.html and the tutorials therein.
Additionally, you can check the Tutorial page at https://docs.epw-code.org/doc/Tutorial.html .
Sincerely,
H. Lee