The electron-phonon vertex calculated by EPW is too large
Posted: Wed Aug 19, 2020 1:40 am
Dear all,
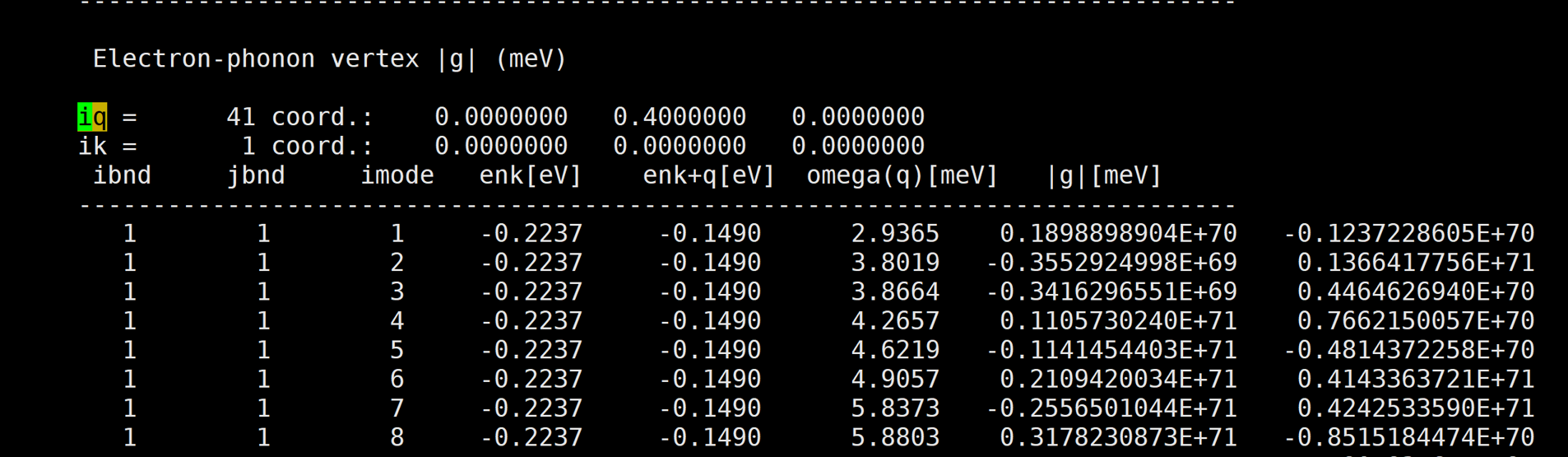
I've calculated naphthalene electron-phonon vertex by qe-6.4.1 and EPW v.5.1.0. But the result is too large.
Here is the result: [img][https://i.loli.net/2020/08/19/PYzZAoGXFMHu9mJ.png]
And the input file i use is
scf.in
-------------------------------------------------------------------------------------
&control
calculation = 'scf'
prefix = 'napht'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
/
&system
ibrav=0,
celldm(1)= 15.32199397
nat=36,
ntyp=2,
ecutwfc=60,
ecutrho=600,
occupations = 'smearing'
smearing = 'gauss'
degauss = 1.0d-9
nbnd = 96
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.081425852 0.016578000 0.320425540
C 0.418563591 0.516578633 0.679561956
C 0.918568223 0.983429349 0.679566194
C 0.581425272 0.483420039 0.320421614
C 0.109536973 0.152891341 0.216967674
C 0.390453781 0.652894793 0.783031949
C 0.890450629 0.847099642 0.783026330
C 0.609537634 0.347109257 0.216971160
C 0.046735358 0.099521927 0.036652394
C 0.453254739 0.599524315 0.963339212
C 0.953256278 0.900476146 0.963345888
C 0.546738013 0.400474080 0.036652955
C 0.073281485 0.236543501 0.926610989
C 0.426711824 0.736536528 0.073385646
C 0.926711917 0.763465068 0.073383309
C 0.573273521 0.263470909 0.926599644
C 0.989266875 0.820195453 0.248086166
C 0.510729800 0.320187579 0.751919324
C 0.010721643 0.179799473 0.751907756
C 0.489267085 0.679804682 0.248087034
H 0.130354682 0.060288819 0.458678341
H 0.369637221 0.560287937 0.541319067
H 0.869636610 0.939711193 0.541317391
H 0.630354818 0.439709201 0.458679729
H 0.179868851 0.304754782 0.271710803
H 0.320123069 0.804753111 0.728283974
H 0.820123570 0.695248369 0.728284733
H 0.679868138 0.195245174 0.271710031
H 0.144899815 0.387460949 0.982909846
H 0.355088380 0.887461819 0.017083198
H 0.855088700 0.612536665 0.017083609
H 0.644903165 0.112531931 0.982912660
H 0.967281381 0.713650483 0.330850532
H 0.532708051 0.213649357 0.669142406
H 0.032708263 0.286351359 0.669142611
H 0.467280791 0.786348134 0.330850337
K_POINTS automatic
4 6 4 0 0 0
CELL_PARAMETERS (alat=15.32199397)
1.070650066 -0.000000900 0.015729322
-0.000000687 0.776651697 -0.000000193
-0.631530124 -0.000000272 0.902091712
------------------------------------------------------------------------------------
nscf_epw.in
------------------------------------------------------------------------------------
&control
calculation = 'nscf'
prefix = 'napht'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
/
&system
ibrav=0,
celldm(1)= 15.32199397
nat=36,
ntyp=2,
ecutwfc=60,
ecutrho=600,
occupations = 'smearing'
smearing = 'gauss'
degauss = 1.0d-9
nbnd = 96
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.081425852 0.016578000 0.320425540
C 0.418563591 0.516578633 0.679561956
C 0.918568223 0.983429349 0.679566194
C 0.581425272 0.483420039 0.320421614
C 0.109536973 0.152891341 0.216967674
C 0.390453781 0.652894793 0.783031949
C 0.890450629 0.847099642 0.783026330
.................
............................
..............................................
CELL_PARAMETERS (alat=15.32199397)
1.070650066 -0.000000900 0.015729322
-0.000000687 0.776651697 -0.000000193
-0.631530124 -0.000000272 0.902091712
K_POINTS crystal
96
0.00000000 0.00000000 0.00000000 1.041667e-02
0.00000000 0.00000000 0.25000000 1.041667e-02
0.00000000 0.00000000 0.50000000 1.041667e-02
0.00000000 0.00000000 0.75000000 1.041667e-02
0.00000000 0.16666667 0.00000000 1.041667e-02
0.00000000 0.16666667 0.25000000 1.041667e-02
0.00000000 0.16666667 0.50000000 1.041667e-02
0.00000000 0.16666667 0.75000000 1.041667e-02
0.00000000 0.33333333 0.00000000 1.041667e-02
0.00000000 0.33333333 0.25000000 1.041667e-02
0.00000000 0.33333333 0.50000000 1.041667e-02
..................................
........................................
...............................................
------------------------------------------------------------------------------------------
epw.in
------------------------------------------------------------------------------------------
--
&inputepw
prefix = 'napht'
amass(1) = 12.01078
amass(2) = 1.007940
outdir = './'
dvscf_dir = './save/'
iverbosity = 1
elph = .true.
kmaps = .false.
epbwrite = .true.
epbread = .false.
! etf_mem = 2 !all the fine Bloch-space el-ph matrix elements are store
epwwrite = .true.
epwread = .false.
nbndsub = 2
nbndskip = 46
bands_skipped = 'exclude_bands = 1:46,49:96'
wannierize = .true.
num_iter = 800
iprint = 2
! dis_win_max = 8
! dis_froz_max= 5.5
!proj(1) = 'Si : sp3'
proj(1) = 'random'
! wdata(1) = 'bands_plot= .true.'
wdata(8) = 'kmesh_tol = 0.00001'
! wdata(2) = 'begin kpoint_path'
! wdata(3) = 'Y 0.5 0 0 G 0 0 0'
! wdata(4) = 'G 0 0 0 B 0 0.5 0'
! wdata(5) = 'B 0 0.5 0 G 0 0 0'
! wdata(6) = 'G 0 0 0 Z 0 0 0.5'
! wdata(7) = 'end kpoint_path'
elecselfen = .false.
phonselfen = .false.
a2f = .false.
fsthick = 20 ! eV
eptemp = 1 ! K
degaussw = 0.01 ! eV
nkf1 = 10
nkf2 = 10
nkf3 = 10
nqf1 = 10
nqf2 = 10
nqf3 = 10
nk1 = 4
nk2 = 6
nk3 = 4
nq1 = 2
nq2 = 3
nq3 = 2
/
8 cartesian
0.000000000000000E+00 0.000000000000000E+00 0.000000000000000E+00
0.806005204269855E-02 -0.129205048000064E-06 -0.548624743748963E+00
0.355229347794158E-06 0.429192821725915E+00 0.378097345350993E-06
0.806040727204635E-02 0.429192692520867E+00 -0.548624365651617E+00
-0.462251719820465E+00 -0.489310279249972E-06 -0.323609985608164E+00
-0.454191667777766E+00 -0.618515327250036E-06 -0.872234729357127E+00
-0.462251364591117E+00 0.429192332415636E+00 -0.323609607510819E+00
-0.454191312548419E+00 0.429192203210588E+00 -0.872234351259782E+00
-------------------------------------------------------------------------------------------------------
epw2.in
-------------------------------------------------------------------------------------------------------
--
&inputepw
prefix = 'napht'
amass(1) = 12.01078
amass(2) = 1.007940
outdir = './'
dvscf_dir = './save/'
iverbosity = 3
elph = .true.
kmaps = .true.
epbwrite = .false.
epbread = .true.
etf_mem = 2 !all the fine Bloch-space el-ph matrix elements are store
epwwrite = .false.
epwread = .true.
lifc = .true.
asr_typ = 'simple'
nbndsub = 2
nbndskip = 46
bands_skipped = 'exclude_bands = 1:46,49:96'
wannierize = .false.
num_iter = 800
iprint = 2
! dis_win_max = 8
! dis_froz_max= 5.5
!proj(1) = 'Si : sp3'
proj(1) = 'random'
wdata(1) = 'bands_plot= .true.'
wdata(9) = 'kmesh_tol = 0.0001'
wdata(2) = 'begin kpoint_path'
wdata(3) = 'Y 0.5 0 0 G 0 0 0'
wdata(4) = 'G 0 0 0 B 0 0.5 0'
wdata(5) = 'B 0 0.5 0 G 0 0 0'
wdata(6) = 'G 0 0 0 Z 0 0 0.5'
wdata(7) = 'end kpoint_path'
wdata(8) = 'bands_num_points = 50'
elecselfen = .false.
phonselfen = .false.
a2f = .false.
band_plot = .false.
fsthick = 20 ! eV
eptemp = 1 ! K
degaussw = 0.01 ! eV
! filqf ='napht_band.qpt'
! filkf ='napht_band.kpt'
nkf1 = 10
nkf2 = 10
nkf3 = 10
nqf1 = 10
nqf2 = 10
nqf3 = 10
nk1 = 4
nk2 = 6
nk3 = 4
nq1 = 2
nq2 = 3
nq3 = 2
/
8 cartesian
0.000000000000000E+00 0.000000000000000E+00 0.000000000000000E+00
0.806005204269855E-02 -0.129205048000064E-06 -0.548624743748963E+00
0.355229347794158E-06 0.429192821725915E+00 0.378097345350993E-06
..................
.............................
............................................
----------------------------------------------------------------------------------------------------------
I don't know where did i go wrong, if you have any idea,please tell me! Thanks a lot!
sincerely,
Jisheng
I've calculated naphthalene electron-phonon vertex by qe-6.4.1 and EPW v.5.1.0. But the result is too large.
Here is the result: [img][https://i.loli.net/2020/08/19/PYzZAoGXFMHu9mJ.png]
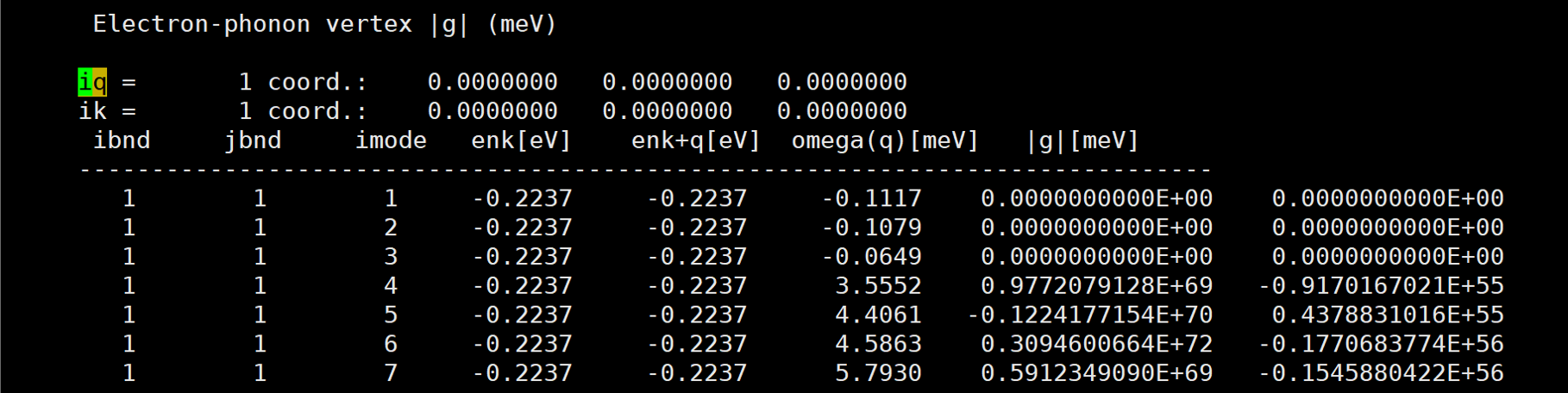{kind=link}
And the input file i use is
scf.in
-------------------------------------------------------------------------------------
&control
calculation = 'scf'
prefix = 'napht'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
/
&system
ibrav=0,
celldm(1)= 15.32199397
nat=36,
ntyp=2,
ecutwfc=60,
ecutrho=600,
occupations = 'smearing'
smearing = 'gauss'
degauss = 1.0d-9
nbnd = 96
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.081425852 0.016578000 0.320425540
C 0.418563591 0.516578633 0.679561956
C 0.918568223 0.983429349 0.679566194
C 0.581425272 0.483420039 0.320421614
C 0.109536973 0.152891341 0.216967674
C 0.390453781 0.652894793 0.783031949
C 0.890450629 0.847099642 0.783026330
C 0.609537634 0.347109257 0.216971160
C 0.046735358 0.099521927 0.036652394
C 0.453254739 0.599524315 0.963339212
C 0.953256278 0.900476146 0.963345888
C 0.546738013 0.400474080 0.036652955
C 0.073281485 0.236543501 0.926610989
C 0.426711824 0.736536528 0.073385646
C 0.926711917 0.763465068 0.073383309
C 0.573273521 0.263470909 0.926599644
C 0.989266875 0.820195453 0.248086166
C 0.510729800 0.320187579 0.751919324
C 0.010721643 0.179799473 0.751907756
C 0.489267085 0.679804682 0.248087034
H 0.130354682 0.060288819 0.458678341
H 0.369637221 0.560287937 0.541319067
H 0.869636610 0.939711193 0.541317391
H 0.630354818 0.439709201 0.458679729
H 0.179868851 0.304754782 0.271710803
H 0.320123069 0.804753111 0.728283974
H 0.820123570 0.695248369 0.728284733
H 0.679868138 0.195245174 0.271710031
H 0.144899815 0.387460949 0.982909846
H 0.355088380 0.887461819 0.017083198
H 0.855088700 0.612536665 0.017083609
H 0.644903165 0.112531931 0.982912660
H 0.967281381 0.713650483 0.330850532
H 0.532708051 0.213649357 0.669142406
H 0.032708263 0.286351359 0.669142611
H 0.467280791 0.786348134 0.330850337
K_POINTS automatic
4 6 4 0 0 0
CELL_PARAMETERS (alat=15.32199397)
1.070650066 -0.000000900 0.015729322
-0.000000687 0.776651697 -0.000000193
-0.631530124 -0.000000272 0.902091712
------------------------------------------------------------------------------------
nscf_epw.in
------------------------------------------------------------------------------------
&control
calculation = 'nscf'
prefix = 'napht'
restart_mode = 'from_scratch'
wf_collect = .false.
pseudo_dir = '/data/home/jspeng/espresso/pseudo'
outdir = './'
tprnfor = .true. !calculate forces
tstress = .true. !calculate stress
/
&system
ibrav=0,
celldm(1)= 15.32199397
nat=36,
ntyp=2,
ecutwfc=60,
ecutrho=600,
occupations = 'smearing'
smearing = 'gauss'
degauss = 1.0d-9
nbnd = 96
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-10
/
ATOMIC_SPECIES
C 12.01078 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.007940d0 H.pbe-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.081425852 0.016578000 0.320425540
C 0.418563591 0.516578633 0.679561956
C 0.918568223 0.983429349 0.679566194
C 0.581425272 0.483420039 0.320421614
C 0.109536973 0.152891341 0.216967674
C 0.390453781 0.652894793 0.783031949
C 0.890450629 0.847099642 0.783026330
.................
............................
..............................................
CELL_PARAMETERS (alat=15.32199397)
1.070650066 -0.000000900 0.015729322
-0.000000687 0.776651697 -0.000000193
-0.631530124 -0.000000272 0.902091712
K_POINTS crystal
96
0.00000000 0.00000000 0.00000000 1.041667e-02
0.00000000 0.00000000 0.25000000 1.041667e-02
0.00000000 0.00000000 0.50000000 1.041667e-02
0.00000000 0.00000000 0.75000000 1.041667e-02
0.00000000 0.16666667 0.00000000 1.041667e-02
0.00000000 0.16666667 0.25000000 1.041667e-02
0.00000000 0.16666667 0.50000000 1.041667e-02
0.00000000 0.16666667 0.75000000 1.041667e-02
0.00000000 0.33333333 0.00000000 1.041667e-02
0.00000000 0.33333333 0.25000000 1.041667e-02
0.00000000 0.33333333 0.50000000 1.041667e-02
..................................
........................................
...............................................
------------------------------------------------------------------------------------------
epw.in
------------------------------------------------------------------------------------------
--
&inputepw
prefix = 'napht'
amass(1) = 12.01078
amass(2) = 1.007940
outdir = './'
dvscf_dir = './save/'
iverbosity = 1
elph = .true.
kmaps = .false.
epbwrite = .true.
epbread = .false.
! etf_mem = 2 !all the fine Bloch-space el-ph matrix elements are store
epwwrite = .true.
epwread = .false.
nbndsub = 2
nbndskip = 46
bands_skipped = 'exclude_bands = 1:46,49:96'
wannierize = .true.
num_iter = 800
iprint = 2
! dis_win_max = 8
! dis_froz_max= 5.5
!proj(1) = 'Si : sp3'
proj(1) = 'random'
! wdata(1) = 'bands_plot= .true.'
wdata(8) = 'kmesh_tol = 0.00001'
! wdata(2) = 'begin kpoint_path'
! wdata(3) = 'Y 0.5 0 0 G 0 0 0'
! wdata(4) = 'G 0 0 0 B 0 0.5 0'
! wdata(5) = 'B 0 0.5 0 G 0 0 0'
! wdata(6) = 'G 0 0 0 Z 0 0 0.5'
! wdata(7) = 'end kpoint_path'
elecselfen = .false.
phonselfen = .false.
a2f = .false.
fsthick = 20 ! eV
eptemp = 1 ! K
degaussw = 0.01 ! eV
nkf1 = 10
nkf2 = 10
nkf3 = 10
nqf1 = 10
nqf2 = 10
nqf3 = 10
nk1 = 4
nk2 = 6
nk3 = 4
nq1 = 2
nq2 = 3
nq3 = 2
/
8 cartesian
0.000000000000000E+00 0.000000000000000E+00 0.000000000000000E+00
0.806005204269855E-02 -0.129205048000064E-06 -0.548624743748963E+00
0.355229347794158E-06 0.429192821725915E+00 0.378097345350993E-06
0.806040727204635E-02 0.429192692520867E+00 -0.548624365651617E+00
-0.462251719820465E+00 -0.489310279249972E-06 -0.323609985608164E+00
-0.454191667777766E+00 -0.618515327250036E-06 -0.872234729357127E+00
-0.462251364591117E+00 0.429192332415636E+00 -0.323609607510819E+00
-0.454191312548419E+00 0.429192203210588E+00 -0.872234351259782E+00
-------------------------------------------------------------------------------------------------------
epw2.in
-------------------------------------------------------------------------------------------------------
--
&inputepw
prefix = 'napht'
amass(1) = 12.01078
amass(2) = 1.007940
outdir = './'
dvscf_dir = './save/'
iverbosity = 3
elph = .true.
kmaps = .true.
epbwrite = .false.
epbread = .true.
etf_mem = 2 !all the fine Bloch-space el-ph matrix elements are store
epwwrite = .false.
epwread = .true.
lifc = .true.
asr_typ = 'simple'
nbndsub = 2
nbndskip = 46
bands_skipped = 'exclude_bands = 1:46,49:96'
wannierize = .false.
num_iter = 800
iprint = 2
! dis_win_max = 8
! dis_froz_max= 5.5
!proj(1) = 'Si : sp3'
proj(1) = 'random'
wdata(1) = 'bands_plot= .true.'
wdata(9) = 'kmesh_tol = 0.0001'
wdata(2) = 'begin kpoint_path'
wdata(3) = 'Y 0.5 0 0 G 0 0 0'
wdata(4) = 'G 0 0 0 B 0 0.5 0'
wdata(5) = 'B 0 0.5 0 G 0 0 0'
wdata(6) = 'G 0 0 0 Z 0 0 0.5'
wdata(7) = 'end kpoint_path'
wdata(8) = 'bands_num_points = 50'
elecselfen = .false.
phonselfen = .false.
a2f = .false.
band_plot = .false.
fsthick = 20 ! eV
eptemp = 1 ! K
degaussw = 0.01 ! eV
! filqf ='napht_band.qpt'
! filkf ='napht_band.kpt'
nkf1 = 10
nkf2 = 10
nkf3 = 10
nqf1 = 10
nqf2 = 10
nqf3 = 10
nk1 = 4
nk2 = 6
nk3 = 4
nq1 = 2
nq2 = 3
nq3 = 2
/
8 cartesian
0.000000000000000E+00 0.000000000000000E+00 0.000000000000000E+00
0.806005204269855E-02 -0.129205048000064E-06 -0.548624743748963E+00
0.355229347794158E-06 0.429192821725915E+00 0.378097345350993E-06
..................
.............................
............................................
----------------------------------------------------------------------------------------------------------
I don't know where did i go wrong, if you have any idea,please tell me! Thanks a lot!
sincerely,
Jisheng
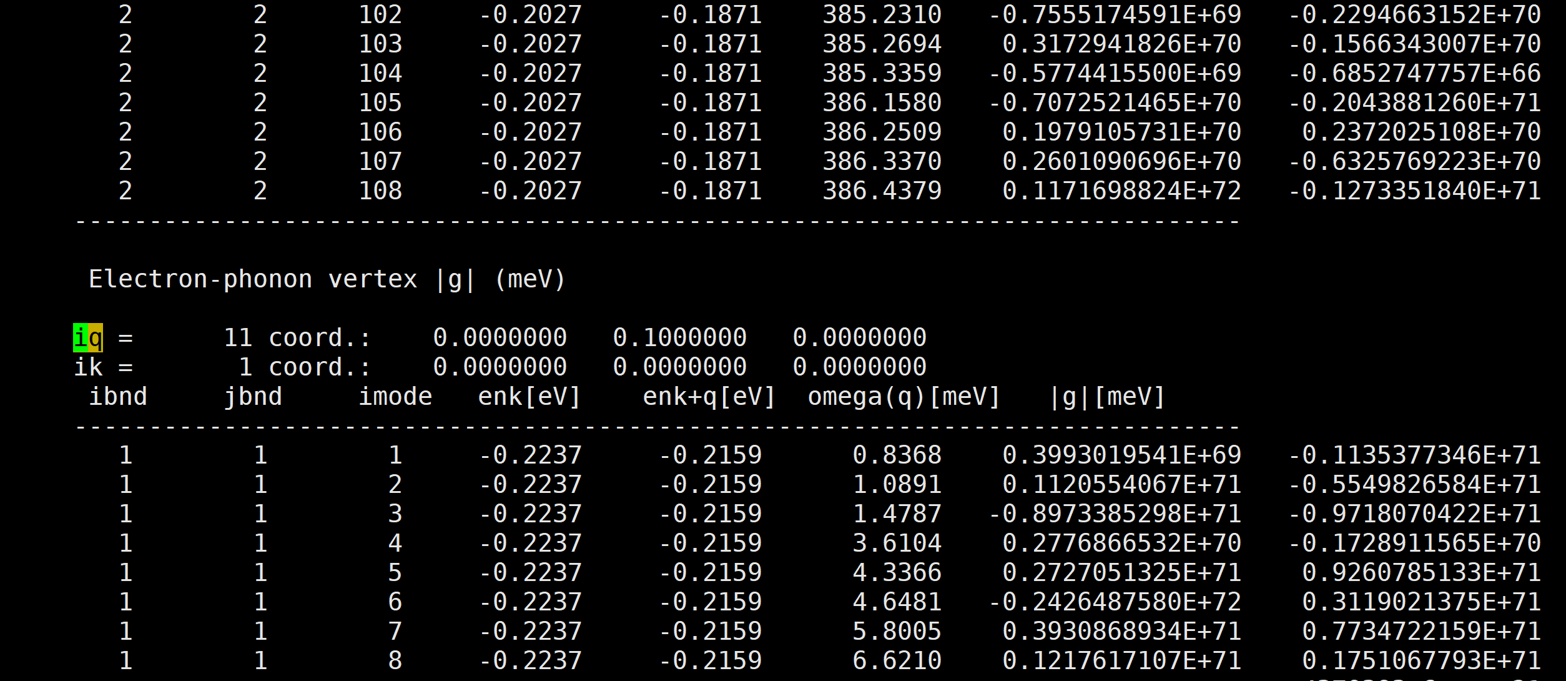{kind=link}
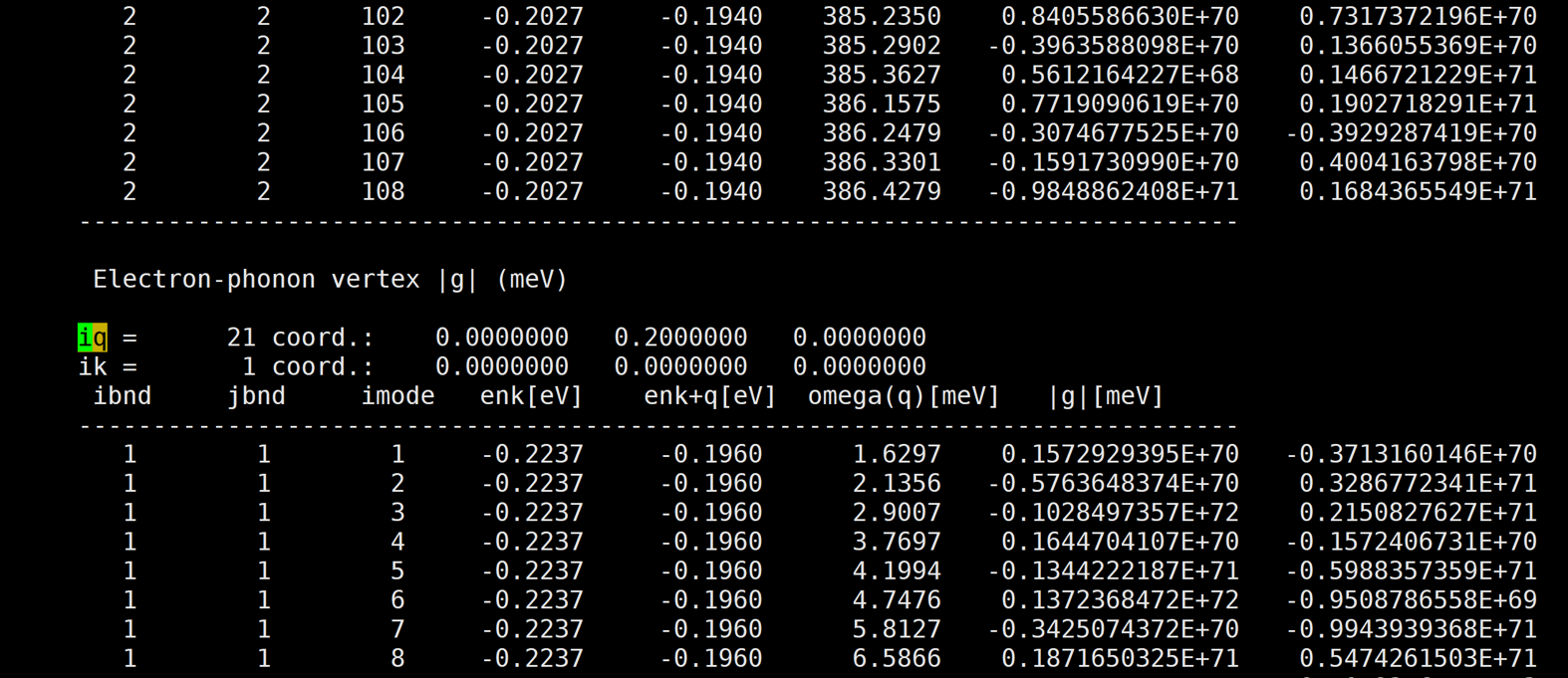{kind=link}
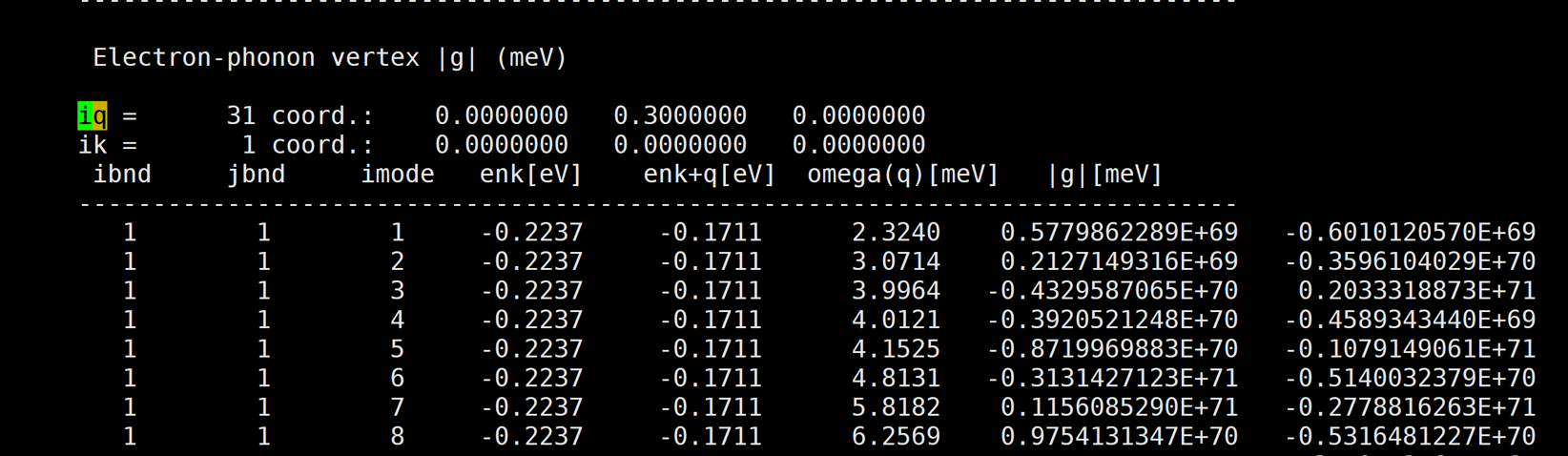{kind=link}
{kind=link}